Clear Sky Science · de
In silico-Entdeckung von Thioglycosid-Analoga als Donorstellen-Inhibitoren der Glycosyltransferase LgtC
Warum das Schwächen von Keimen besser sein kann als ihr Abtöten
Krankenhäuser weltweit kämpfen mit Infektionen durch Bakterien, die auf viele Antibiotika nicht mehr ansprechen. Anstatt diese Mikroben direkt zu töten, verfolgen einige Forscher eine andere Strategie: sie gezielt entwaffnen, sodass unser Immunsystem und vorhandene Medikamente den Rest erledigen können. Diese Studie nutzt Computermodelle, um nach neuen kleinen Molekülen, sogenannten Thioglycosiden, zu suchen, die ein wichtiges bakterielles Enzym blockieren und gefährliche Gram‑negative Erreger deutlich weniger krankmachend machen könnten.
Das Problem harter äußerer Schilde
Viele der besorgniserregendsten Bakterien, etwa Arten von Neisseria und andere Gram‑negative Keime, sind von einer komplexen Außenhülle aus Zuckermolekülen umgeben. Ein Teil dieser Hülle, bekannt als Lipooligosaccharid, hilft ihnen, der Immunabwehr zu entgehen und gegenüber Antibiotika resistent zu sein. Der Aufbau dieses Schutzmantels erfordert eine Reihe spezialisierter Enzyme. Eines davon, LgtC genannt, hängt eine Zuckergruppe namens Galactose an die wachsende Außenschicht. Wird LgtC blockiert, bleibt die Oberflächenstruktur unvollständig und die Bakterien werden verwundbarer. Da menschliche Zellen LgtC nicht nutzen, ist es ein attraktives Ziel für Wirkstoffe, die Krankheitserreger schwächen könnten, ohne uns zu schaden.
Mit Computern den chemischen Raum durchsuchen
Die Forscher konzentrierten sich auf eine Familie zuckerähnlicher Moleküle, die Thioglycoside, bei denen ein Schwefelatom die im natürlichen Zucker übliche Sauerstoffverbindung ersetzt. Frühere Arbeiten zeigten, dass zwei solche Verbindungen, FucSBn und BacSBn, die bakterielle Zuckerassemblierung stören können. In dieser Untersuchung durchsuchte das Team die große PubChem‑Datenbank nach Molekülen, die diesen beiden "metabolischen Lockvögeln" ähnlich sind. Es blieben nur Kandidaten übrig, die nach üblichen Regeln zur Arzneimitteleignung sowie vorhergesagten Resorptions‑ und Sicherheitsprofilen für eine Weiterentwicklung als Medikamente geeignet erschienen. Dieser Filter schränkte die Auswahl auf 18 vielversprechende Analoga ein, die klein genug, nicht zu fett- oder wasserliebend und bei therapeutischen Dosen voraussichtlich nicht hoch toxisch sind.
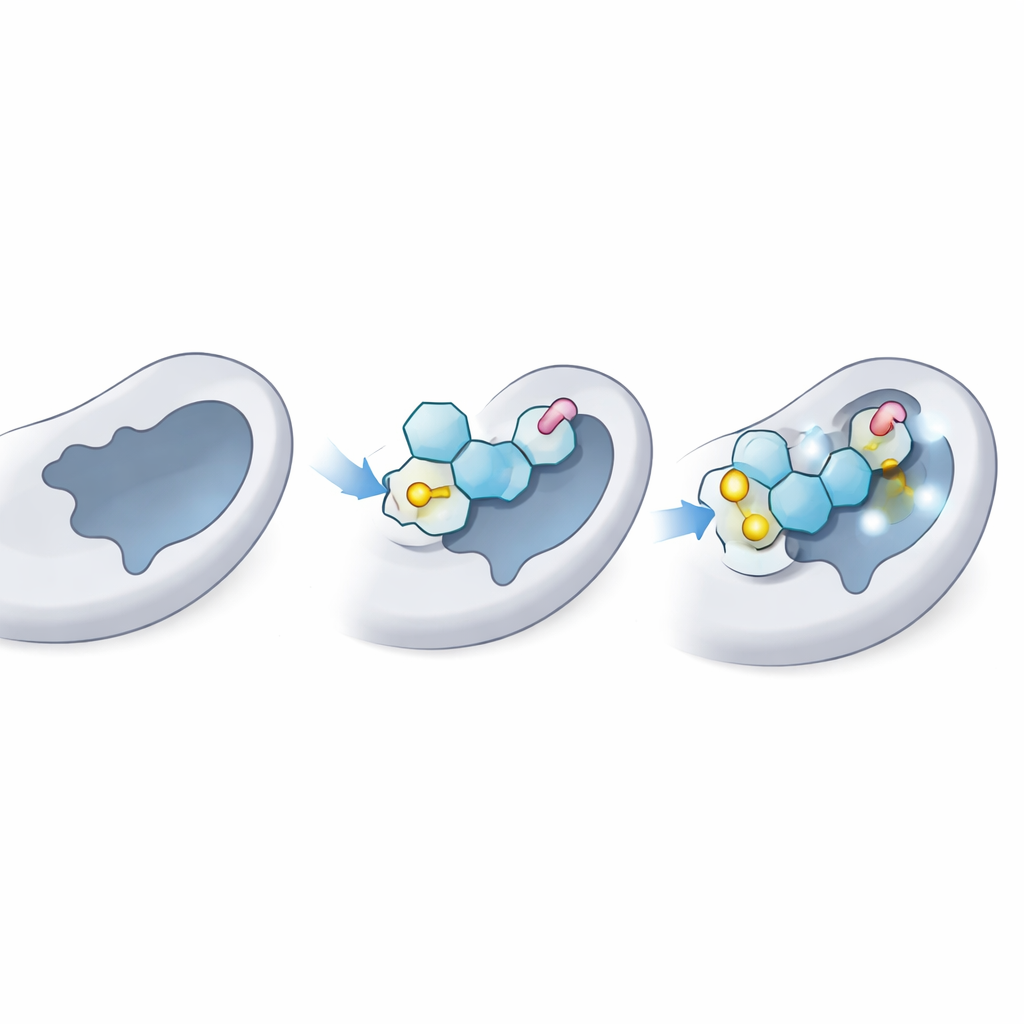
Prüfen, wie gut die Kandidaten ins Enzym passen
Als Nächstes verwendeten die Wissenschaftler molekulares Docking, eine Art virtuelles Schlüssel‑Schloss‑Testverfahren, um zu sehen, wie eng jedes Thioglycosid in die Donorstelle von LgtC passen könnte — die Tasche, in der normalerweise der natürliche Zucker-Donor bindet. Zuerst bestätigten sie, dass ihre Methode den natürlichen Donor korrekt in die bekannte 3D‑Struktur von LgtC "redocken" kann und so experimentelle Daten reproduziert. Dann dockten sie alle 18 Analoga tausendfach. Mehrere, insbesondere drei mit den Bezeichnungen C‑5, C‑14 und C‑18, zeigten durchgängig stärkere vorhergesagte Bindung als der natürliche Donor, was darauf hindeutet, dass sie effektiv um dieselbe Stelle am Enzym konkurrieren könnten.
Die Interaktion in Bewegung beobachten
Docking liefert ein Standbild; das Team führte daher 100‑Nanosekunden-Molekulardynamik‑Simulationen durch, um zu sehen, wie sich das Enzym und jedes Ligand im Laufe der Zeit in einer virtuellen wässrigen Umgebung verhalten. Diese Simulationen verfolgen, wie stark das Komplex schwankt, wie kompakt das Protein bleibt und welche Kontakte bestehen bleiben. Die besten Thioglycoside hielten eine stabile Pose in der Donor‑Tasche bei, mit nur moderater Bewegung vergleichbar zum natürlichen Zucker. Sie bewahrten wichtige Wasserstoffbrücken und Nahkontakte zu denselben Ankerresten, die normalerweise den realen Donor festhalten, und gewannen zusätzliche stabilisierende Wechselwirkungen durch ihre Schwefelbindungen und aromatischen „Schwänze“. Gesamtgestalt und Flexibilität des Proteins blieben in gesunden Grenzen, was darauf hindeutet, dass das Enzym nicht verzerrt, sondern lediglich blockiert wurde.
Was das für zukünftige Behandlungen bedeutet
Zusammenfassend deuten virtuelle Screenings, Docking‑Werte und lange Simulationen auf eine kleine Gruppe von Thioglycosid‑Gerüsten — insbesondere C‑5, C‑14 und C‑18 — als starke Kandidaten, um als kompetitive Blocker von LgtC zu fungieren. Einfach ausgedrückt scheinen diese Moleküle lange genug und fest genug in der Zuckerbindungsstelle des Enzyms sitzen zu können, um zu verhindern, dass die echten Bausteine eindringen. Das sollte den Aufbau der bakteriellen Schutzschicht stören und die Mikroben schwächen, ohne sie unbedingt direkt abzutöten. Die Arbeit ist vollständig rechnerisch und muss noch in Enzym‑ und Zellexperimenten bestätigt werden, bietet aber eine klare Roadmap für Chemiker: Diese Thioglycoside liefern Ausgangsblaupausen für die Entwicklung der nächsten Generation antivirulenter Wirkstoffe, die helfen könnten, multiresistente Gram‑negative Infektionen in den Griff zu bekommen.
Zitation: Sierra-Hernández, O., Saurith-Coronell, O., Alcázar, J.J. et al. In silico discovery of thioglycoside analogues as donor-site inhibitors of glycosyltransferase LgtC. Sci Rep 16, 13807 (2026). https://doi.org/10.1038/s41598-026-43638-x
Schlüsselwörter: Antivirulenz, Gramnegative Bakterien, Glycosyltransferase LgtC, Thioglycosid-Inhibitoren, in silico Wirkstoffsuche