Clear Sky Science · pt
Identificação e engenharia de proteases potyvirais altamente funcionais em células usando modelos de coevolução
Tesouras moleculares mais precisas para a ciência e a medicina
A biologia moderna frequentemente depende de pequenas “tesouras” moleculares — proteases — que cortam proteínas exatamente no ponto certo. Esses cortes podem ativar ou desligar processos celulares, ajudar a purificar proteínas produzidas em laboratório ou impulsionar circuitos genéticos projetados. No entanto, para uma grande família de proteases virais amplamente usada em pesquisa, seu poder de corte e precisão nunca foram totalmente mapeados. Este estudo apresenta um método orientado por dados para descobrir, prever e redesenhar essas proteases para que funcionem melhor e com mais seletividade dentro de células humanas, abrindo caminho para biotecnologia mais limpa e até destruição direcionada de células prejudiciais.
Por que os cortadores de proteínas virais importam
O trabalho foca nas proteases NIa, enzimas de potyvírus que infectam plantas. Um membro dessa família, a protease do vírus do mosaico do tabaco (TEVp), é uma ferramenta padrão em engenharia de proteínas porque reconhece uma sequência muito específica de sete aminoácidos e corta em um ponto definido. Mas a TEVp é apenas uma entre mais de 3.800 proteases relacionadas cujas capacidades permanecem em grande parte inexploradas. Se os cientistas pudessem entender sistematicamente qual protease corta qual sequência, e como mudanças sutis na sequência alteram a atividade, poderiam trocar por “tesouras” melhores para o trabalho de laboratório, construir circuitos sintéticos mais complexos em células e projetar proteases que respondam apenas a mutações associadas a doenças.
Aprendendo com os padrões da natureza
Para enfrentar isso, os autores reuniram 3.817 pares naturais de proteases potyvirais e seus alvos correspondentes, incluindo não só o núcleo de sete letras do sítio de corte, mas também os aminoácidos ao redor. Em seguida, construíram o ProSSpeC, um modelo computacional que busca padrões de coevolução entre a protease e seu substrato — posições que mudam em conjunto ao longo da evolução para preservar um bom encaixe. Usando um esquema de pontuação inspirado na física, o modelo atribui a cada par protease–substrato uma pontuação de especificidade: quanto mais favorável (mais negativa) a pontuação, maior a probabilidade de o par cortar eficientemente. Ao subtrair padrões que surgem de similaridade geral em vez de interação real, o ProSSpeC foca nas características que realmente importam para reconhecer e cortar o sítio correto.
Colocando previsões à prova em células humanas
A equipe então perguntou se esses números realmente preveem o comportamento em células vivas. Eles projetaram um ensaio fluorescente em células humanas onde um corte bem-sucedido reconstitui uma proteína vermelha fluorescente dividida, produzindo um sinal brilhante que é normalizado por um repórter verde. Testando dezenas de combinações protease–substrato, descobriram que pares com pontuações ProSSpeC mais fortes tendiam a gerar sinais mais intensos, e que proteases naturais geralmente preferiam suas próprias sequências-alvo nativas. Em 225 combinações de 15 proteases e 15 substratos, as pontuações computacionais se correlacionaram bem com a fluorescência medida e distinguiram com precisão pares que cortavam daqueles que não cortavam, mesmo quando apenas o motivo central de sete aminoácidos foi considerado.
Ajustando cortes um bloco de construção de cada vez
Como o ProSSpeC opera em resolução de único aminoácido, os autores o usaram para explorar o que acontece quando apenas um bloco de construção no alvo é alterado. Para múltiplos pares protease–substrato, preveram mutações que deveriam aumentar ou diminuir o corte, depois construíram essas variantes e mediram sua atividade. Mudanças na pontuação do modelo acompanharam de perto as variações na fluorescência, confirmando que ele pode prever como um único resíduo altera o desempenho. O modelo também destacou a importância do contexto mais amplo em torno do sítio central: substituir um motivo repetido simples pela vizinhança natural de 20 aminoácidos frequentemente aumentou a clivagem várias vezes, apoiando a ideia de que resíduos flanqueadores, moldados pela evolução, ajustam com precisão o reconhecimento.
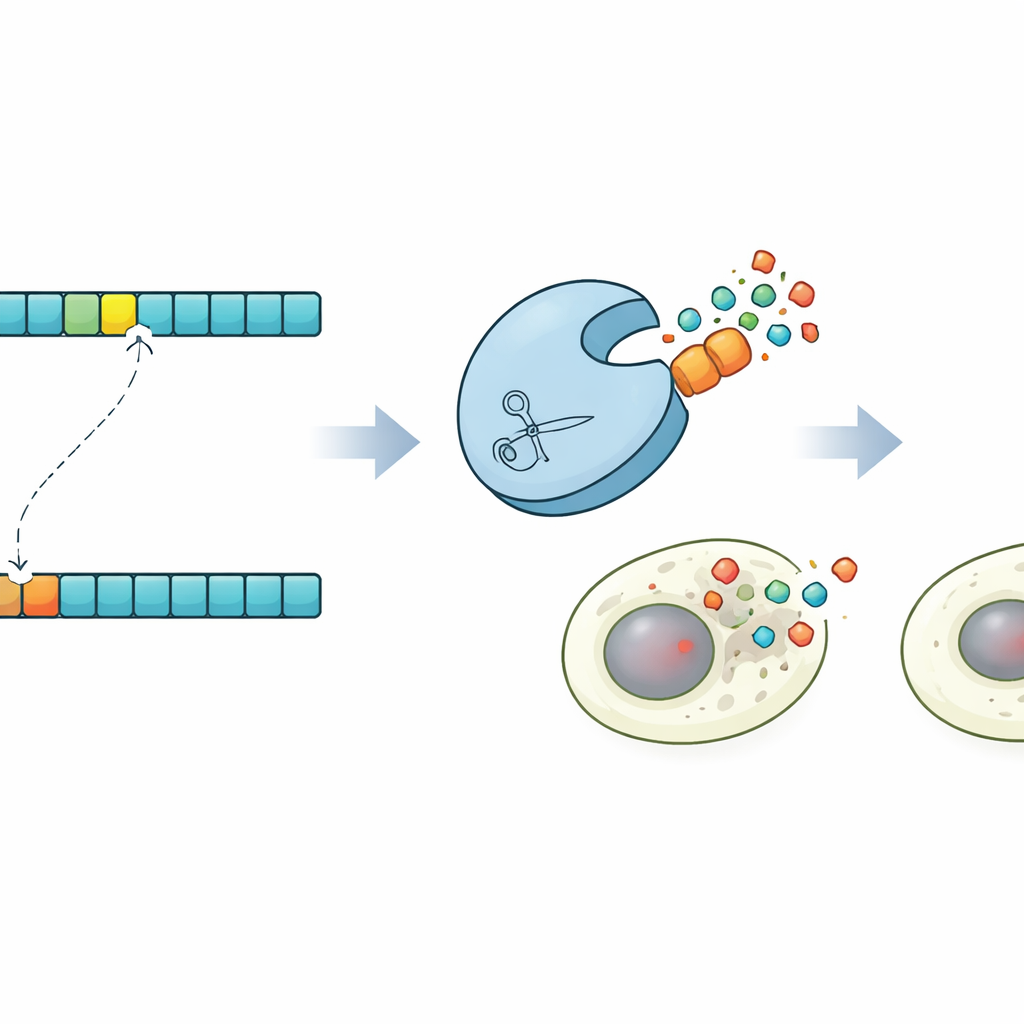
Morte celular programada sob demanda
Para mostrar o que esse controle torna possível, os pesquisadores projetaram uma demonstração impressionante em células humanas. Usando suas previsões, identificaram uma protease que cortaria uma versão sutilmente mutada de um sítio alvo, mas deixaria a versão original intacta. Em seguida, construíram versões da Caspase-3, uma executora chave da morte celular programada, que carregavam ou o sítio normal ou o mutado. Em populações celulares mistas, a protease projetada ativou seletivamente a Caspase-3 apenas em células com a sequência mutante, desencadeando sua apoptose enquanto poupava células vizinhas. Esse circuito de “sinpoptose” mostra que o design de proteases guiado por coevolução pode ser usado para detectar diferenças de um único aminoácido e convertê-las em decisões de vida ou morte para células.
O que isso significa daqui para frente
Para não especialistas, a mensagem principal é que os autores transformaram pistas evolutivas dispersas em sequências de proteínas em uma ferramenta prática de projeto para tesouras moleculares. O ProSSpeC não só encontra proteases que superam um cavalo de batalha de laboratório como a TEVp, mas também explica quais contatos entre enzima e alvo importam mais, mesmo quando atuam à distância. Embora haja limites para sequências muito diferentes de qualquer coisa encontrada na natureza, o modelo já oferece aos pesquisadores uma maneira de navegar e redesenhar milhares de proteases virais para cortes mais limpos e especificidades personalizadas. A longo prazo, tais ferramentas podem ajudar a construir terapias celulares mais inteligentes, diagnósticos melhores e sistemas programáveis que editem redes de proteínas dentro de células com a mesma precisão que o CRISPR trouxe para o DNA.
Citação: Lim Suan, M.B., Ziegler, C., Syed, Z. et al. Identification and engineering of highly functional potyviral proteases in cells using co-evolutionary models. Nat Commun 17, 3257 (2026). https://doi.org/10.1038/s41467-026-69961-5
Palavras-chave: engenharia de protease, modelagem coevolutiva, biologia sintética, morte celular programada, especificidade de proteínas