Clear Sky Science · en
Identification and engineering of highly functional potyviral proteases in cells using co-evolutionary models
Sharper molecular scissors for science and medicine
Modern biology often relies on tiny molecular "scissors"—proteases—that cut proteins at just the right spot. These cuts can turn cellular processes on or off, help purify lab-made proteins, or drive engineered genetic circuits. Yet for one large family of viral proteases widely used in research, their cutting power and precision have never been fully mapped. This study introduces a data-driven way to discover, predict, and redesign these proteases so they work better and more selectively inside human cells, opening doors to cleaner biotechnology and even targeted destruction of harmful cells.
Why viral protein cutters matter
The work focuses on NIa proteases, enzymes from plant-infecting potyviruses. One member of this family, the tobacco etch virus protease (TEVp), is a staple tool in protein engineering because it recognizes a very specific seven–amino-acid sequence and cuts at a defined point. But TEVp is just one of more than 3,800 related proteases whose abilities remain mostly unexplored. If scientists could systematically understand which protease cuts which sequence, and how subtle sequence changes alter activity, they could swap in better "scissors" for lab work, build more complex synthetic circuits in cells, and design proteases that respond only to disease-linked mutations.
Learning from nature’s patterns
To tackle this, the authors gathered 3,817 natural pairs of potyviral proteases and their matching protein targets, including not just the core seven-letter cutting site but also the surrounding amino acids. They then built ProSSpeC, a computational model that looks for co-evolving patterns between the protease and its substrate—positions that change together over evolution to preserve a good fit. Using a physics-inspired scoring scheme, the model assigns each protease–substrate pair a specificity score: the more favorable (more negative) the score, the more likely the pair is to cut efficiently. By subtracting patterns that arise from general similarity rather than real interaction, ProSSpeC homes in on features that truly matter for recognizing and cutting the right site.
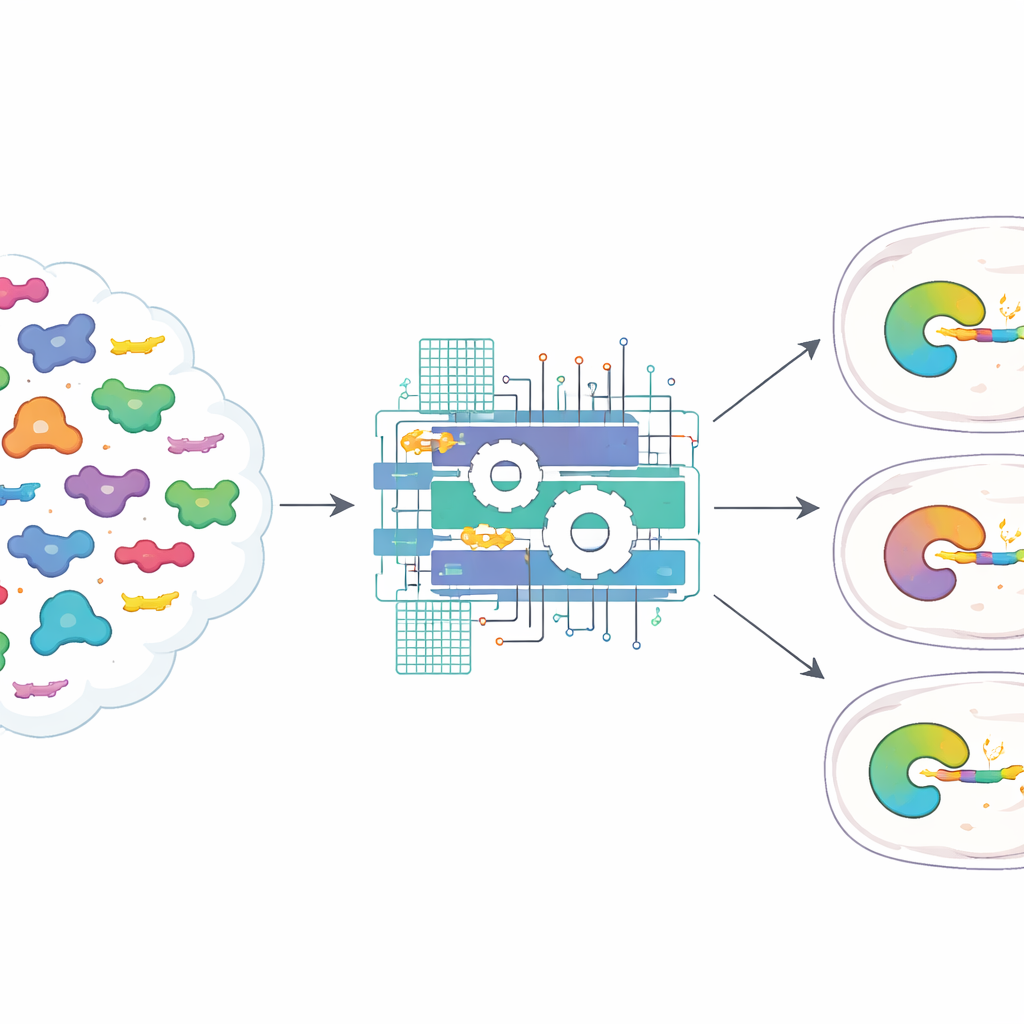
Putting predictions to the test in human cells
The team next asked whether these numbers actually predict behavior in living cells. They designed a fluorescent assay in human cells where a successful cut reassembles a split red fluorescent protein, producing a bright signal that is normalized to a green reporter. Testing dozens of protease–substrate combinations, they found that pairs with stronger ProSSpeC scores tended to give brighter signals, and that natural proteases generally preferred their own native target sequences. Across 225 combinations of 15 proteases and 15 substrates, the computational scores correlated well with measured fluorescence and accurately distinguished cutting from non-cutting pairs, even when only the seven–amino-acid core motif was considered.
Tuning cuts one building block at a time
Because ProSSpeC operates at single–amino-acid resolution, the authors used it to explore what happens when just one building block in the target sequence is changed. For multiple protease–substrate pairs, they predicted mutations that should either boost or weaken cutting, then built these variants and measured their activity. Changes in the model’s score closely tracked changes in fluorescence, confirming that it can foresee how a single residue alters performance. The model also highlighted the importance of the broader sequence context around the core site: replacing a simple repeated motif with the natural 20–amino-acid neighborhood often boosted cleavage several-fold, supporting the idea that flanking residues, shaped by evolution, fine-tune recognition.
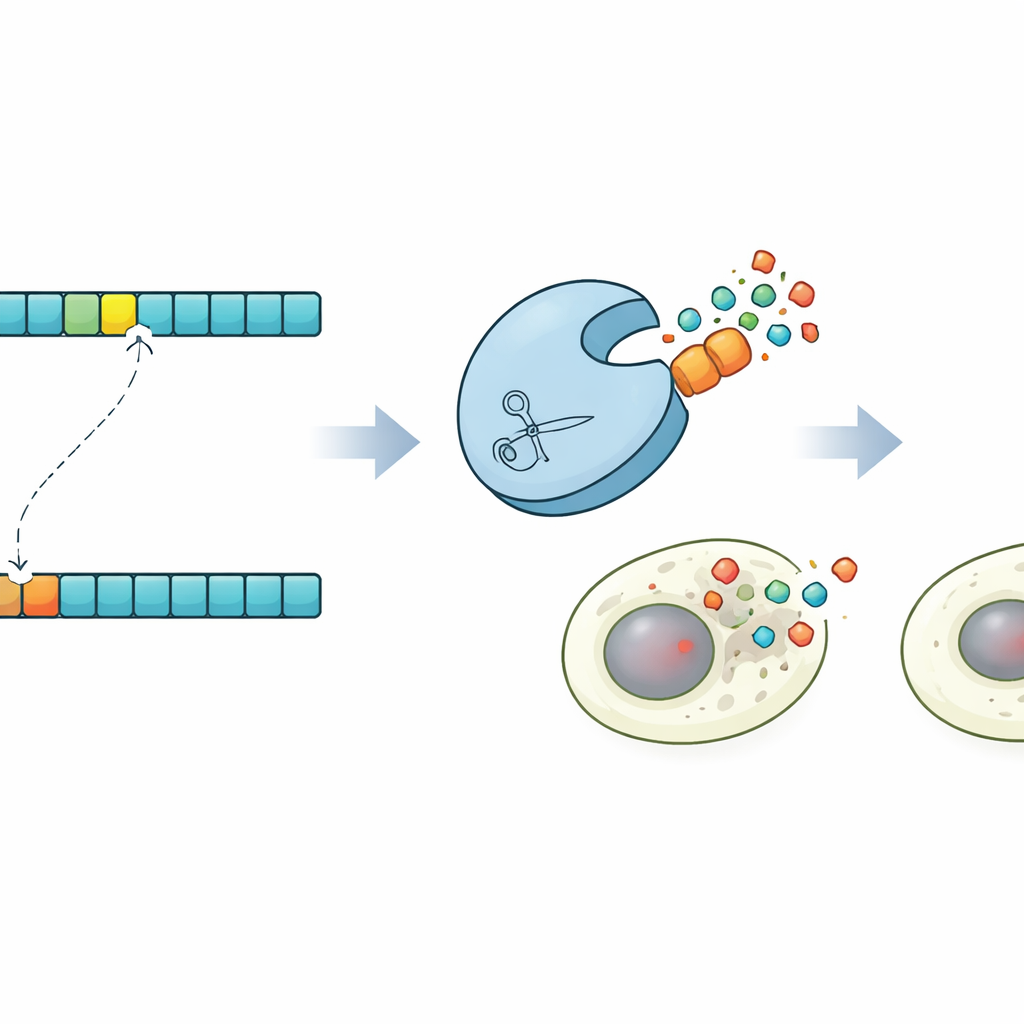
Programmed cell death on command
To showcase what this control makes possible, the researchers engineered a striking demonstration in human cells. Using their predictions, they identified a protease that would cut a subtly mutated version of a target site but leave the original version untouched. They then built versions of Caspase-3, a key executioner of programmed cell death, that carry either the normal or mutated site. In mixed cell populations, the engineered protease selectively activated Caspase-3 only in cells with the mutant sequence, triggering their apoptosis while sparing neighboring cells. This "synpoptosis" circuit shows that co-evolution–guided protease design can be harnessed to detect single–amino-acid differences and convert them into life-or-death decisions for cells.
What this means going forward
For non-specialists, the key message is that the authors have turned scattered evolutionary hints in protein sequences into a practical design tool for molecular scissors. ProSSpeC not only finds proteases that outperform a standard lab workhorse like TEVp, but also explains which contacts between enzyme and target matter most, even when they act at a distance. While there are limits for sequences far from anything found in nature, the model already gives researchers a way to browse and redesign thousands of viral proteases for cleaner cuts and custom specificities. In the long run, such tools could help build smarter cell therapies, better diagnostics, and programmable systems that edit protein networks inside cells with the same precision that CRISPR brought to DNA.
Citation: Lim Suan, M.B., Ziegler, C., Syed, Z. et al. Identification and engineering of highly functional potyviral proteases in cells using co-evolutionary models. Nat Commun 17, 3257 (2026). https://doi.org/10.1038/s41467-026-69961-5
Keywords: protease engineering, co-evolutionary modeling, synthetic biology, programmed cell death, protein specificity