Clear Sky Science · fr
Identification et ingénierie de protéases potyvirales hautement fonctionnelles dans les cellules en utilisant des modèles de co-évolution
Des ciseaux moléculaires plus précis pour la science et la médecine
La biologie moderne repose souvent sur de minuscules « ciseaux » moléculaires — les protéases — qui coupent les protéines au bon endroit. Ces coupures peuvent activer ou désactiver des processus cellulaires, aider à purifier des protéines produites en laboratoire ou piloter des circuits génétiques conçus. Pourtant, pour une grande famille de protéases virales largement utilisées en recherche, leur puissance de coupe et leur précision n’ont jamais été complètement cartographiées. Cette étude présente une approche fondée sur les données pour découvrir, prédire et redesigner ces protéases afin qu’elles fonctionnent mieux et plus sélectivement dans les cellules humaines, ouvrant la voie à une biotechnologie plus propre et même à la destruction ciblée de cellules nocives.
Pourquoi les coupeurs de protéines viraux comptent
Le travail se concentre sur les protéases NIa, des enzymes issues de potyvirus infectant les plantes. Un membre de cette famille, la protéase du virus de l’étch tobaccique (TEVp), est un outil incontournable en ingénierie des protéines car elle reconnaît une séquence très spécifique de sept acides aminés et coupe en un point défini. Mais la TEVp n’est qu’un des plus de 3 800 protéases apparentées dont les capacités restent pour la plupart inexplorées. Si les scientifiques pouvaient comprendre systématiquement quelle protéase coupe quelle séquence, et comment de subtiles variations de séquence modifient l’activité, ils pourraient remplacer les « ciseaux » existants par de meilleurs outils pour le laboratoire, construire des circuits synthétiques cellulaires plus complexes et concevoir des protéases qui ne répondent qu’à des mutations liées à la maladie.
Apprendre des motifs de la nature
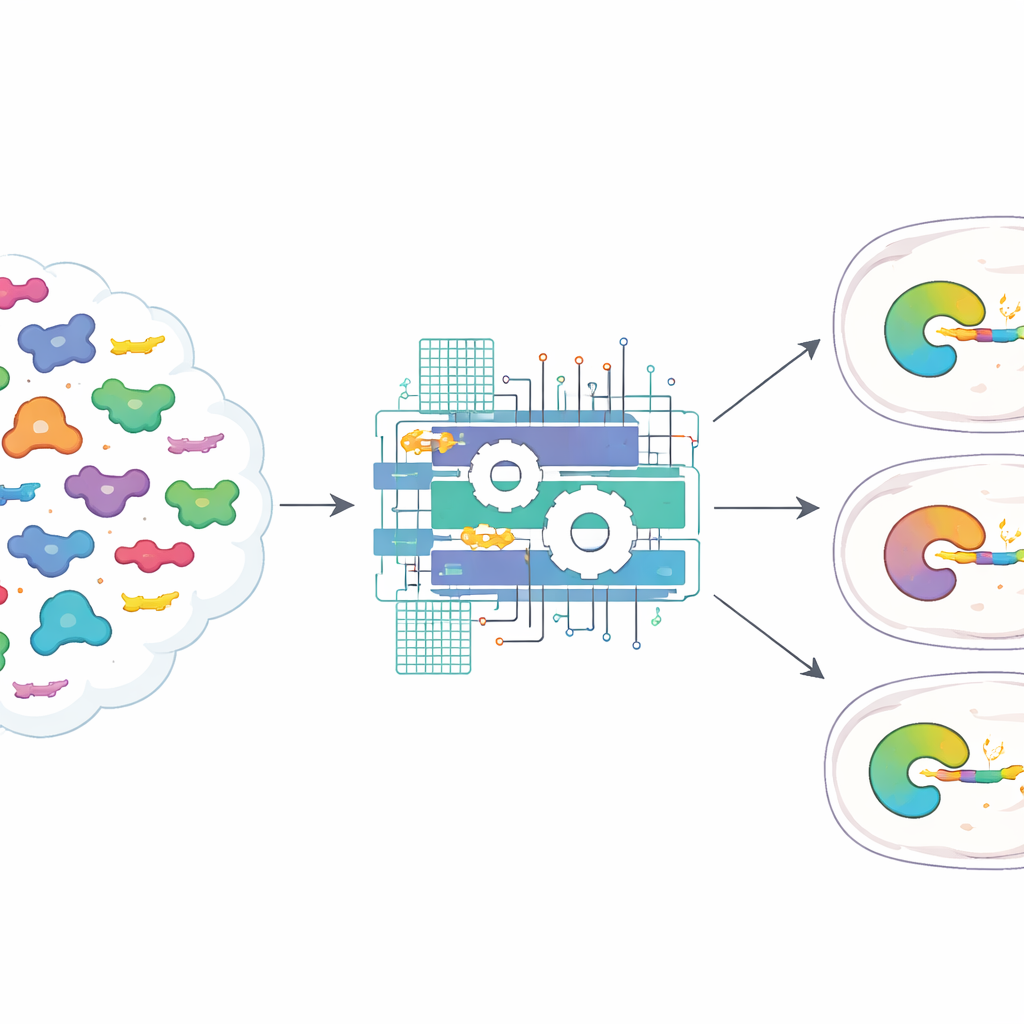
Pour s’attaquer à ce défi, les auteurs ont rassemblé 3 817 paires naturelles de protéases potyvirales et de leurs substrats correspondants, en incluant non seulement le site d’action central de sept lettres mais aussi les acides aminés environnants. Ils ont ensuite construit ProSSpeC, un modèle informatique qui recherche des motifs co-évolutifs entre la protéase et son substrat — des positions qui évoluent de concert pour préserver une bonne affinité. En utilisant un schéma de score inspiré de la physique, le modèle attribue à chaque paire protéase–substrat un score de spécificité : plus le score est favorable (plus négatif), plus la paire a de chances de couper efficacement. En soustrayant les motifs résultant d’une similarité générale plutôt que d’une interaction réelle, ProSSpeC identifie les caractéristiques qui comptent vraiment pour reconnaître et couper le bon site.
Mettre les prédictions à l’épreuve dans des cellules humaines
L’équipe a ensuite vérifié si ces valeurs prédites reflétaient le comportement dans des cellules vivantes. Ils ont conçu un test fluorescent dans des cellules humaines où une coupure réussie réassemble une protéine fluorescente rouge scindée, produisant un signal brillant normalisé par un témoin vert. En testant des dizaines de combinaisons protéase–substrat, ils ont constaté que les paires avec des scores ProSSpeC plus forts donnaient généralement des signaux plus lumineux, et que les protéases naturelles préféraient en général leurs propres séquences cibles natives. Sur 225 combinaisons de 15 protéases et 15 substrats, les scores informatiques corrélaient bien avec la fluorescence mesurée et distinguaient avec précision les paires coupantes des paires non coupantes, même lorsque seul le motif central de sept acides aminés était considéré.
Ajuster les coupures, un bloc de construction à la fois
Parce que ProSSpeC opère à la résolution d’un seul acide aminé, les auteurs l’ont utilisé pour explorer ce qui se passe lorsqu’un seul bloc de construction dans la séquence cible est modifié. Pour plusieurs paires protéase–substrat, ils ont prédit des mutations qui devraient soit augmenter soit diminuer la coupure, puis ont construit ces variantes et mesuré leur activité. Les variations du score du modèle suivaient de près les changements de fluorescence, confirmant qu’il peut prévoir comment un seul résidu modifie la performance. Le modèle a également mis en évidence l’importance du contexte séquentiel élargi autour du site central : remplacer un motif répétitif simple par le voisinage naturel de 20 acides aminés augmentait souvent la clivage de plusieurs fois, soutenant l’idée que les résidus flanquants, façonnés par l’évolution, affinent la reconnaissance.
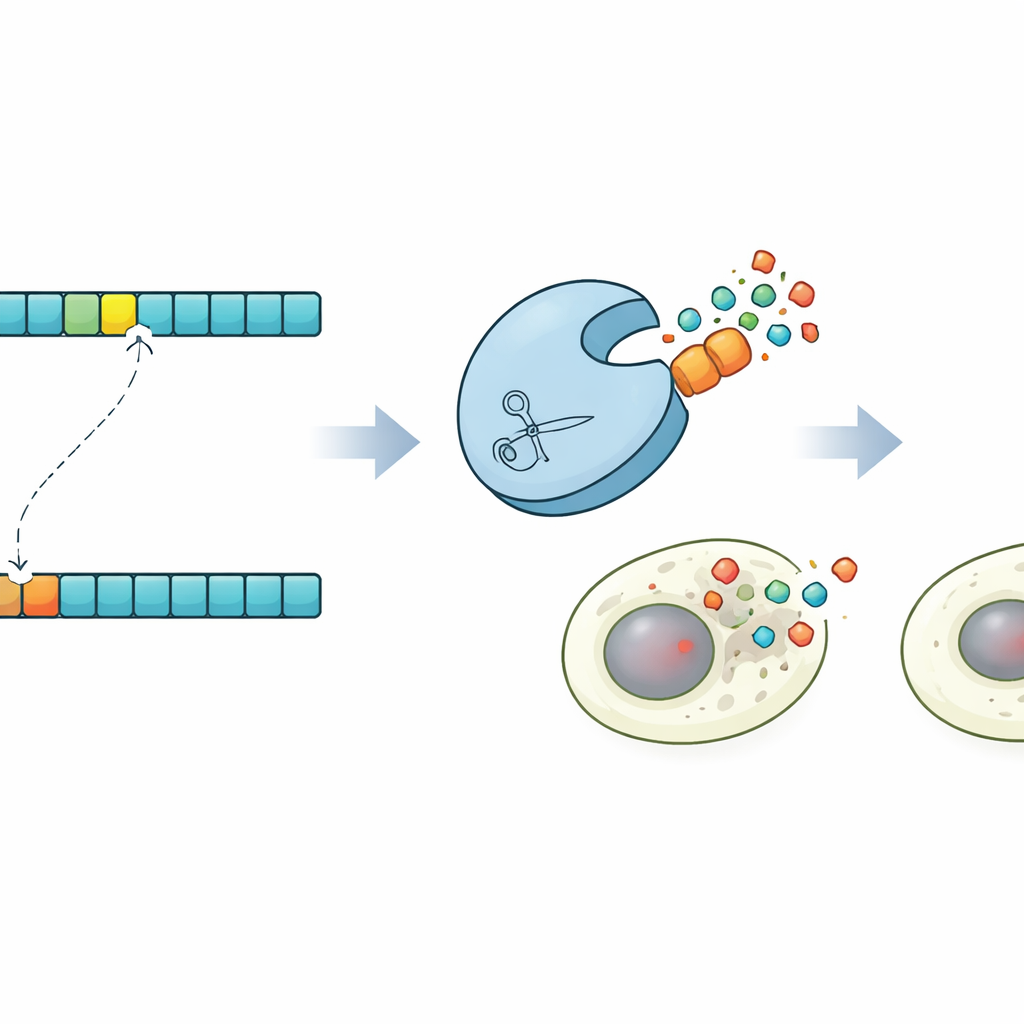
Mort cellulaire programmée sur commande
Pour illustrer ce que ce contrôle rend possible, les chercheurs ont conçu une démonstration frappante dans des cellules humaines. À l’aide de leurs prédictions, ils ont identifié une protéase capable de couper une version subtilement mutée d’un site cible tout en épargnant la version originale. Ils ont ensuite construit des versions de la Caspase-3, un effecteur clé de la mort cellulaire programmée, portant soit le site normal soit le site muté. Dans des populations cellulaires mixtes, la protéase conçue a activé sélectivement la Caspase-3 uniquement dans les cellules portant la séquence mutante, déclenchant leur apoptose tout en épargnant les cellules voisines. Ce circuit de « synpoptose » montre que la conception de protéases guidée par la co-évolution peut détecter des différences d’un seul acide aminé et les convertir en décisions de vie ou de mort pour les cellules.
Ce que cela signifie pour l’avenir
Pour les non-spécialistes, le message principal est que les auteurs ont transformé des indices évolutifs dispersés dans des séquences protéiques en un outil de conception pratique pour des ciseaux moléculaires. ProSSpeC ne se contente pas de trouver des protéases qui surpassent un standard de laboratoire comme la TEVp, il explique aussi quels contacts entre l’enzyme et la cible importent le plus, même lorsqu’ils agissent à distance. S’il existe des limites pour des séquences très éloignées de tout ce que la nature a produit, le modèle offre déjà aux chercheurs un moyen d’explorer et de redesigner des milliers de protéases virales pour obtenir des coupures plus propres et des spécificités sur mesure. À long terme, de tels outils pourraient aider à concevoir des thérapies cellulaires plus intelligentes, de meilleurs diagnostics et des systèmes programmables qui éditent les réseaux protéiques à l’intérieur des cellules avec la même précision que CRISPR a apportée à l’ADN.
Citation: Lim Suan, M.B., Ziegler, C., Syed, Z. et al. Identification and engineering of highly functional potyviral proteases in cells using co-evolutionary models. Nat Commun 17, 3257 (2026). https://doi.org/10.1038/s41467-026-69961-5
Mots-clés: ingénierie des protéases, modélisation co-évolutive, biologie synthétique, mort cellulaire programmée, spécificité des protéines