Clear Sky Science · es
Identificación e ingeniería de proteasas potyvirales altamente funcionales en células usando modelos de coevolución
Tijeras moleculares más precisas para la ciencia y la medicina
La biología moderna a menudo depende de pequeñas «tijeras» moleculares —proteasas— que cortan proteínas en el lugar exacto. Estos cortes pueden activar o desactivar procesos celulares, ayudar a purificar proteínas producidas en el laboratorio o impulsar circuitos genéticos diseñados. Sin embargo, para una gran familia de proteasas virales ampliamente utilizadas en investigación, su potencia y precisión de corte nunca se han cartografiado por completo. Este estudio presenta un enfoque basado en datos para descubrir, predecir y rediseñar estas proteasas para que funcionen mejor y con más selectividad dentro de células humanas, abriendo puertas a una biotecnología más limpia e incluso a la destrucción dirigida de células dañinas.
Por qué importan los cortadores de proteínas virales
El trabajo se centra en las proteasas NIa, enzimas de potyvirus que infectan plantas. Un miembro de esta familia, la proteasa del virus del estriado del tabaco (TEVp), es una herramienta básica en ingeniería de proteínas porque reconoce una secuencia muy específica de siete aminoácidos y corta en un punto definido. Pero TEVp es solo una de más de 3.800 proteasas relacionadas cuyas capacidades siguen siendo en gran parte inexploradas. Si los científicos pudieran comprender sistemáticamente qué proteasa corta qué secuencia y cómo cambios sutiles en la secuencia alteran la actividad, podrían reemplazar por unas «tijeras» mejores para el trabajo de laboratorio, construir circuitos sintéticos más complejos en células y diseñar proteasas que respondan solo a mutaciones vinculadas a enfermedades.
Aprendiendo de los patrones de la naturaleza
Para abordar esto, los autores reunieron 3.817 pares naturales de proteasas potyvirales y sus dianas proteicas coincidentes, incluyendo no solo el motivo central de siete letras donde ocurre el corte, sino también los aminoácidos circundantes. Luego construyeron ProSSpeC, un modelo computacional que busca patrones de coevolución entre la proteasa y su sustrato —posiciones que cambian juntas a lo largo de la evolución para conservar un buen encaje. Usando un esquema de puntuación inspirado en la física, el modelo asigna a cada par proteasa–sustrato una puntuación de especificidad: cuanto más favorable (más negativa) es la puntuación, más probable es que el par corte de forma eficiente. Al restar los patrones que surgen de la similitud general en lugar de la interacción real, ProSSpeC se centra en las características que verdaderamente importan para reconocer y cortar el sitio correcto.
Comprobando las predicciones en células humanas
El equipo preguntó a continuación si estas cifras realmente predicen el comportamiento en células vivas. Diseñaron un ensayo fluorescente en células humanas en el que un corte exitoso vuelve a ensamblar una proteína roja fluorescente dividida, produciendo una señal brillante que se normaliza frente a un reportero verde. Al probar docenas de combinaciones proteasa–sustrato, encontraron que los pares con puntuaciones ProSSpeC más fuertes tendían a dar señales más intensas, y que las proteasas naturales generalmente preferían sus propias secuencias diana nativas. En 225 combinaciones de 15 proteasas y 15 sustratos, las puntuaciones computacionales se correlacionaron bien con la fluorescencia medida y distinguieron con precisión pares que cortan de los que no cortan, incluso cuando se consideró solo el motivo central de siete aminoácidos.
Ajustando los cortes un bloque a la vez
Como ProSSpeC opera a resolución de un solo aminoácido, los autores lo utilizaron para explorar qué ocurre cuando se cambia solo un bloque de construcción en la secuencia diana. Para múltiples pares proteasa–sustrato, predijeron mutaciones que deberían aumentar o debilitar el corte, luego construyeron estas variantes y midieron su actividad. Los cambios en la puntuación del modelo siguieron de cerca los cambios en la fluorescencia, confirmando que puede prever cómo un único residuo altera el rendimiento. El modelo también destacó la importancia del contexto secuencial más amplio alrededor del sitio central: reemplazar un motivo repetido simple por el vecindario natural de 20 aminoácidos a menudo aumentó la escisión varias veces, apoyando la idea de que los residuos flanqueantes, moldeados por la evolución, afinan el reconocimiento.
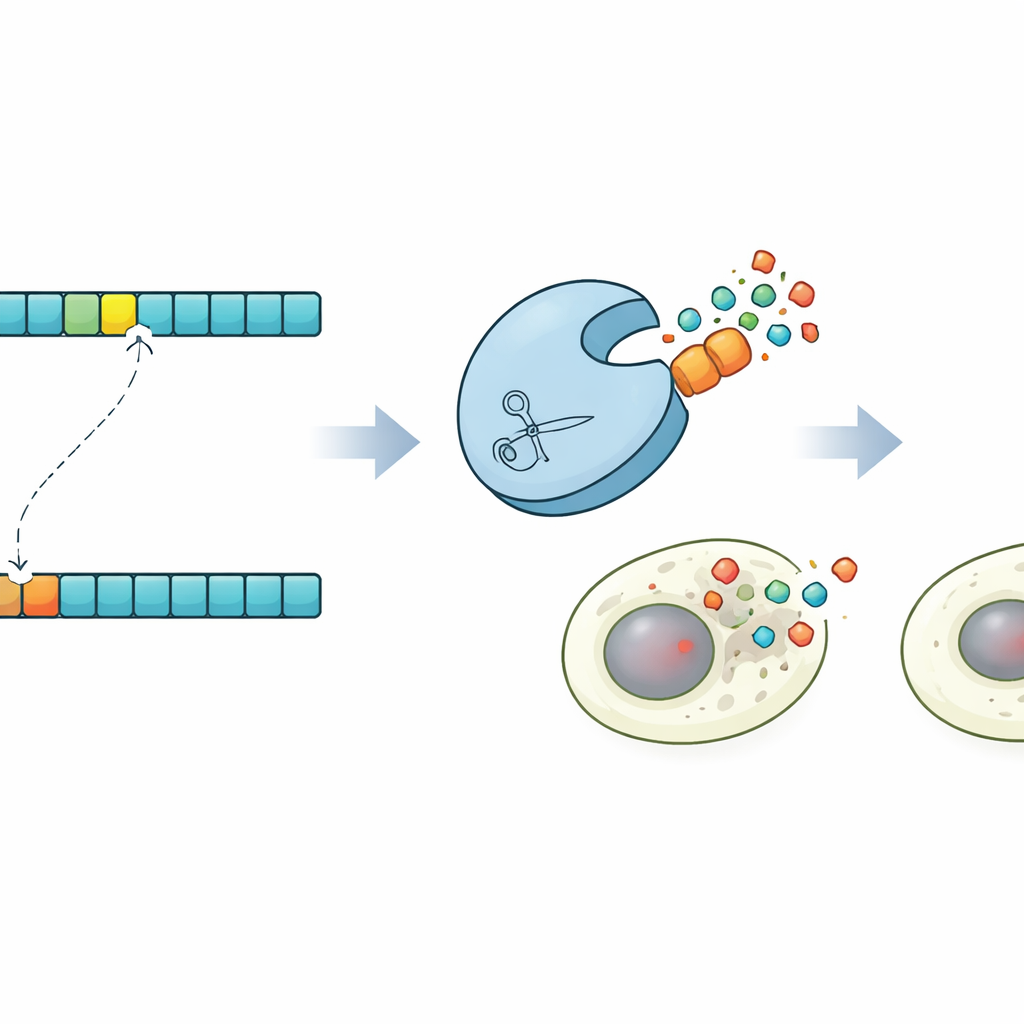
Muerte celular programada bajo demanda
Para mostrar lo que permite este control, los investigadores diseñaron una demostración llamativa en células humanas. Usando sus predicciones, identificaron una proteasa que cortaría una versión sutilmente mutada de un sitio diana pero dejaría intacta la versión original. A continuación, construyeron versiones de la Caspasa-3, un ejecutor clave de la muerte celular programada, que portaban ya sea el sitio normal o el mutado. En poblaciones celulares mixtas, la proteasa diseñada activó selectivamente la Caspasa-3 solo en las células con la secuencia mutante, desencadenando su apoptosis mientras preservaba las células vecinas. Este circuito de «sinpoptofosis» muestra que el diseño de proteasas guiado por la coevolución puede aprovecharse para detectar diferencias de un solo aminoácido y convertirlas en decisiones de vida o muerte para las células.
Qué implica esto en el futuro
Para quienes no son especialistas, el mensaje clave es que los autores han convertido indicios evolutivos dispersos en secuencias de proteínas en una herramienta práctica de diseño para tijeras moleculares. ProSSpeC no solo encuentra proteasas que superan a una herramienta estándar de laboratorio como TEVp, sino que también explica qué contactos entre enzima y diana importan más, incluso cuando actúan a distancia. Aunque hay límites para secuencias muy alejadas de lo que se encuentra en la naturaleza, el modelo ya ofrece a los investigadores una forma de explorar y rediseñar miles de proteasas virales para cortes más limpios y especificidades personalizadas. A largo plazo, tales herramientas podrían ayudar a construir terapias celulares más inteligentes, mejores diagnósticos y sistemas programables que editen redes de proteínas dentro de las células con la misma precisión que CRISPR logró con el ADN.
Cita: Lim Suan, M.B., Ziegler, C., Syed, Z. et al. Identification and engineering of highly functional potyviral proteases in cells using co-evolutionary models. Nat Commun 17, 3257 (2026). https://doi.org/10.1038/s41467-026-69961-5
Palabras clave: ingeniería de proteasas, modelado de coevolución, biología sintética, muerte celular programada, especificidad de proteínas