Clear Sky Science · pl
Identyfikacja i inżynieria wysoko funkcjonalnych proteaz potywirusowych w komórkach przy użyciu modeli koewolucyjnych
Bardziej precyzyjne molekularne nożyczki dla nauki i medycyny
Współczesna biologia często polega na drobnych molekularnych „nożyczkach” — proteazach — które przecinają białka w dokładnie odpowiednim miejscu. Takie cięcia mogą włączać lub wyłączać procesy komórkowe, pomagać w oczyszczaniu białek wytwarzanych w laboratorium lub napędzać zaprojektowane obwody genetyczne. Jednak w przypadku dużej rodziny wirusowych proteaz często wykorzystywanych w badaniach, ich siła cięcia i precyzja nie zostały dotąd w pełni zmapowane. Niniejsze badanie przedstawia oparte na danych podejście do odkrywania, przewidywania i przeprojektowywania tych proteaz, tak aby działały lepiej i bardziej selektywnie w komórkach ludzkich, otwierając drogę do czystszej biotechnologii, a nawet ukierunkowanej eliminacji szkodliwych komórek.
Dlaczego wirusowe „nożyce” mają znaczenie
Praca koncentruje się na proteazach NIa, enzymach pochodzących z potywirusów atakujących rośliny. Jeden z członków tej rodziny, proteaza wirusa bruzdkowego tytoniu (TEVp), jest powszechnie stosowanym narzędziem w inżynierii białek, ponieważ rozpoznaje bardzo specyficzny siedmioaminokwasowy fragment i tnie w zdefiniowanym miejscu. Jednak TEVp to tylko jeden z ponad 3 800 spokrewnionych proteaz, których możliwości pozostają w dużej mierze nieprzebadane. Gdyby naukowcy mogli systematycznie zrozumieć, która proteaza rozcina który sekwencję i jak subtelne zmiany w sekwencji wpływają na aktywność, mogliby wymieniać „nożyce” na lepsze do prac laboratoryjnych, budować bardziej złożone syntetyczne obwody w komórkach i projektować proteazy reagujące tylko na mutacje związane z chorobą.
Nauka z wzorców natury
Aby to osiągnąć, autorzy zgromadzili 3 817 naturalnych par proteaza–substrat z potywirusów, obejmujących nie tylko rdzeń siedmiu znaków miejsca cięcia, lecz także otaczające aminokwasy. Następnie zbudowali ProSSpeC, model komputerowy poszukujący koewoluujących wzorców między proteazą a jej substratem — pozycji, które zmieniają się razem w toku ewolucji, aby zachować dobre dopasowanie. Korzystając z fizykopodobnego schematu oceniania, model przypisuje każdej parze proteaza–substrat wynik specyficzności: im bardziej korzystny (bardziej ujemny) wynik, tym większe prawdopodobieństwo efektywnego cięcia. Poprzez odjęcie wzorców wynikających z ogólnego podobieństwa, a nie rzeczywistej interakcji, ProSSpeC skupia się na cechach, które naprawdę mają znaczenie dla rozpoznawania i przecinania właściwego miejsca.
Sprawdzanie przewidywań w komórkach ludzkich
Zespół zapytał następnie, czy te wartości rzeczywiście przewidują zachowanie w żywych komórkach. Zaprojektowali fluorescencyjny test w komórkach ludzkich, w którym udane cięcie ponownie łączy rozdzielone czerwone białko fluorescencyjne, generując jasny sygnał normalizowany względem zielonego markera. Testując kilkadziesiąt kombinacji proteaza–substrat, stwierdzili, że pary z lepszymi wynikami ProSSpeC zwykle dawały silniejszy sygnał, a proteazy naturalne generalnie preferowały własne natywne sekwencje docelowe. Wśród 225 kombinacji 15 proteaz i 15 substratów wyniki obliczeniowe dobrze korelowały z mierzonymi wartościami fluorescencji i skutecznie rozróżniały pary tnące od nietnących, nawet gdy brano pod uwagę jedynie siedmioaminokwasowy rdzeń motywu.
Dostrajanie cięć po jednym elemencie
Ponieważ ProSSpeC działa z rozdzielczością pojedynczego aminokwasu, autorzy użyli go do zbadania, co się dzieje, gdy zmieni się tylko jeden element w sekwencji docelowej. Dla wielu par proteaza–substrat przewidzieli mutacje, które powinny albo zwiększyć, albo osłabić cięcie, a następnie zbudowali te warianty i zmierzyli ich aktywność. Zmiany w wyniku modelu ściśle odzwierciedlały zmiany we fluorescencji, potwierdzając, że potrafi on przewidzieć, jak pojedyncze reszty wpływają na działanie. Model podkreślił także wagę szerszego kontekstu sekwencji wokół rdzenia: zastąpienie prostego powtarzalnego motywu naturalnym, 20‑aminokwasowym sąsiedztwem często zwiększało szybkość cięcia kilkukrotnie, co wspiera pogląd, że reszty flankingowe, kształtowane przez ewolucję, precyzują rozpoznawanie.
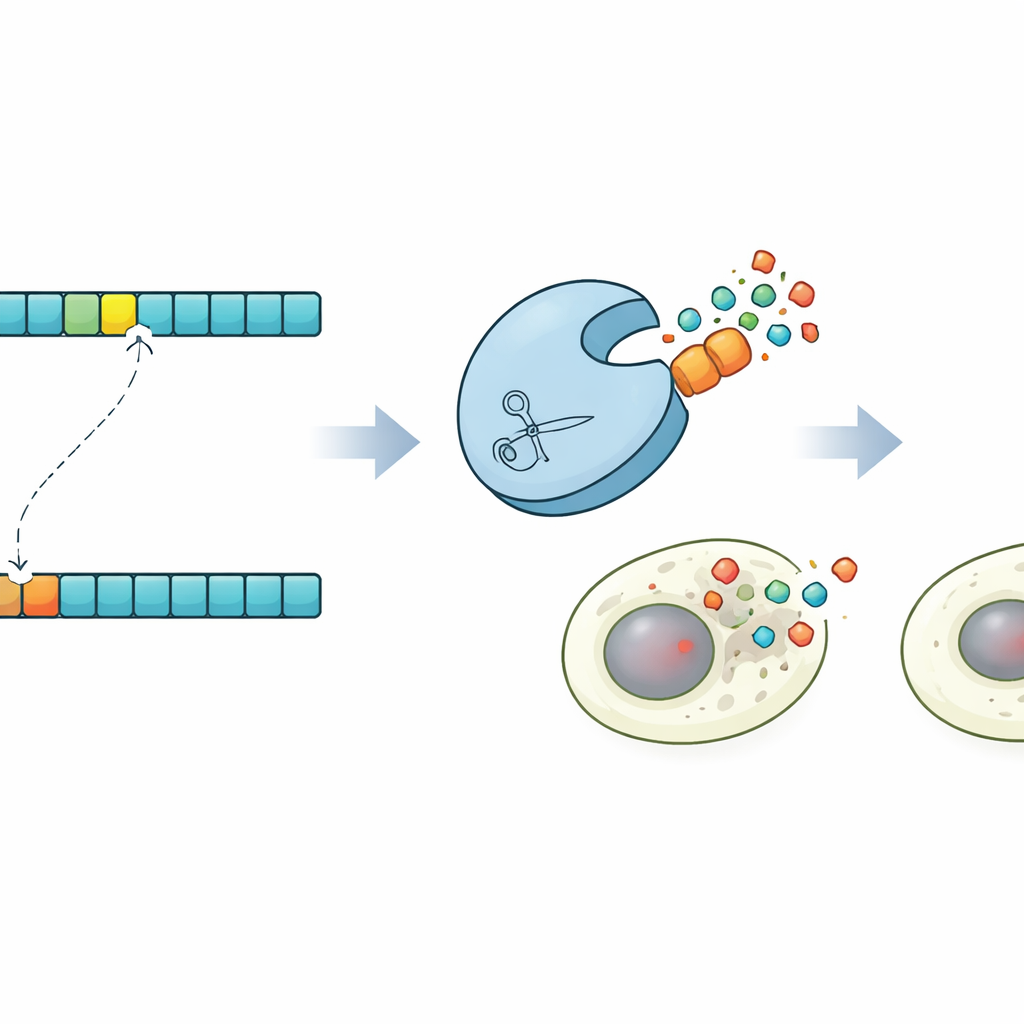
Zaprogramowana śmierć komórki na zawołanie
Aby zademonstrować możliwości tej kontroli, badacze zaprojektowali efektowny pokaz w komórkach ludzkich. Korzystając ze swoich przewidywań, zidentyfikowali proteazę, która ma ciąć subtelnie zmutowaną wersję miejsca docelowego, pozostawiając nietkniętą wersję oryginalną. Następnie zbudowali warianty Kaspazy-3, kluczowego egzekutora zaprogramowanej śmierci komórki, niosące albo normalne, albo zmutowane miejsce. W mieszanych populacjach komórek zaprojektowana proteaza selektywnie aktywowała Kaspazę-3 tylko w komórkach z sekwencją mutantową, wywołując ich apoptozę i oszczędzając sąsiednie komórki. Ten układ „synpoptosis” pokazuje, że projektowanie proteaz kierowane koewolucją można wykorzystać do wykrywania różnic pojedynczych aminokwasów i przekształcania ich w decyzje o życiu lub śmierci dla komórek.
Co to oznacza na przyszłość
Dla osób spoza dziedziny kluczowy przekaz jest taki, że autorzy przekształcili rozproszone ewolucyjne wskazówki w sekwencjach białek w praktyczne narzędzie projektowe dla molekularnych nożyczek. ProSSpeC nie tylko znajduje proteazy, które przewyższają standardowe laboratoryjne narzędzie, takie jak TEVp, lecz także wyjaśnia, które kontakty między enzymem a celem mają największe znaczenie, nawet gdy działają z dystansu. Choć istnieją ograniczenia dla sekwencji bardzo różniących się od czegokolwiek występującego w naturze, model już teraz daje badaczom sposób przeglądania i przeprojektowywania tysięcy wirusowych proteaz w celu uzyskania czyściejszych cięć i niestandardowej specyficzności. W dłuższej perspektywie takie narzędzia mogą pomóc w tworzeniu inteligentniejszych terapii komórkowych, lepszych diagnostyk i programowalnych systemów, które edytują sieci białkowe w komórkach z tą samą precyzją, jaką CRISPR przyniósł dla DNA.
Cytowanie: Lim Suan, M.B., Ziegler, C., Syed, Z. et al. Identification and engineering of highly functional potyviral proteases in cells using co-evolutionary models. Nat Commun 17, 3257 (2026). https://doi.org/10.1038/s41467-026-69961-5
Słowa kluczowe: inżynieria proteaz, modelowanie koewolucyjne, biologia syntetyczna, zaprogramowana śmierć komórki, specyficzność białka