Clear Sky Science · it
Identificazione e ingegnerizzazione di proteasi potyvirali altamente funzionali nelle cellule usando modelli co-evolutivi
Forbici molecolari più affilate per la scienza e la medicina
La biologia moderna si affida spesso a piccole “forbici” molecolari — le proteasi — che tagliano le proteine nel punto giusto. Questi tagli possono attivare o disattivare processi cellulari, aiutare a purificare proteine prodotte in laboratorio o guidare circuiti genetici ingegnerizzati. Tuttavia, per una vasta famiglia di proteasi virali ampiamente usata nella ricerca, la potenza di taglio e la precisione non sono mai state completamente mappate. Questo studio introduce un approccio guidato dai dati per scoprire, prevedere e riprogettare queste proteasi in modo che funzionino meglio e con maggiore selettività nelle cellule umane, aprendo la strada a biotecnologie più pulite e persino alla distruzione mirata di cellule dannose.
Perché i tagli proteici virali sono importanti
Il lavoro si concentra sulle proteasi NIa, enzimi provenienti da potyvirus che infettano le piante. Un membro di questa famiglia, la proteasi del Tobacco Etch Virus (TEVp), è uno strumento fondamentale nell’ingegneria delle proteine perché riconosce una sequenza molto specifica di sette aminoacidi e taglia in un punto definito. Ma TEVp è solo una delle oltre 3.800 proteasi affini le cui capacità restano per lo più inesplorate. Se gli scienziati potessero capire sistematicamente quale proteasi taglia quale sequenza e come sottili cambiamenti di sequenza alterano l’attività, potrebbero sostituire le “forbici” con versioni migliori per il lavoro di laboratorio, costruire circuiti sintetici più complessi nelle cellule e progettare proteasi che rispondono solo a mutazioni associate a malattie.
Imparare dai modelli della natura
Per affrontare il problema, gli autori hanno raccolto 3.817 coppie naturali di proteasi potyvirali e dei loro bersagli proteici corrispondenti, includendo non solo il nucleo di sette lettere dove avviene il taglio, ma anche gli aminoacidi circostanti. Hanno quindi costruito ProSSpeC, un modello computazionale che cerca schemi co-evolutivi tra la proteasi e il suo substrato — posizioni che cambiano insieme nel corso dell’evoluzione per mantenere un buon incastro. Utilizzando uno schema di scoring ispirato alla fisica, il modello assegna a ciascuna coppia proteasi–substrato un punteggio di specificità: più favorevole (più negativo) è il punteggio, più probabile è che la coppia tagli efficientemente. Sottraendo schemi che emergono dalla somiglianza generale piuttosto che da vere interazioni, ProSSpeC si concentra sulle caratteristiche che contano davvero per riconoscere e tagliare il sito corretto.
Mettere alla prova le previsioni nelle cellule umane
Il gruppo ha poi verificato se questi valori predittivi corrispondono al comportamento nelle cellule viventi. Hanno progettato un saggio fluorescente in cellule umane in cui un taglio riuscito riassembla una proteina rossa fluorescente divisa, producendo un segnale intenso normalizzato su un marcatore verde. Testando dozzine di combinazioni proteasi–substrato, hanno trovato che le coppie con punteggi ProSSpeC più forti tendevano a dare segnali più luminosi e che le proteasi naturali preferivano generalmente le proprie sequenze bersaglio native. Su 225 combinazioni di 15 proteasi e 15 substrati, i punteggi computazionali correlavano bene con la fluorescenza misurata e distinguevano con precisione le coppie che tagliano da quelle che non tagliano, anche quando si considerava solo il motivo centrale di sette aminoacidi.
Regolare i tagli un blocco alla volta
Poiché ProSSpeC opera a risoluzione di singolo aminoacido, gli autori lo hanno usato per esplorare cosa succede cambiando un solo componente nella sequenza bersaglio. Per più coppie proteasi–substrato, hanno previsto mutazioni che avrebbero dovuto aumentare o ridurre il taglio, quindi hanno costruito queste varianti e misurato la loro attività. Le variazioni nel punteggio del modello riflettevano da vicino le variazioni nella fluorescenza, confermando che può prevedere come una singola residua modifichi le prestazioni. Il modello ha inoltre evidenziato l’importanza del contesto sequenziale più ampio attorno al sito centrale: sostituire un motivo ripetuto semplice con il vicinato naturale di 20 aminoacidi spesso aumentava la scissione anche di diversi ordini di grandezza, a sostegno dell’idea che i residui flangiatrici, plasmati dall’evoluzione, affinano il riconoscimento.
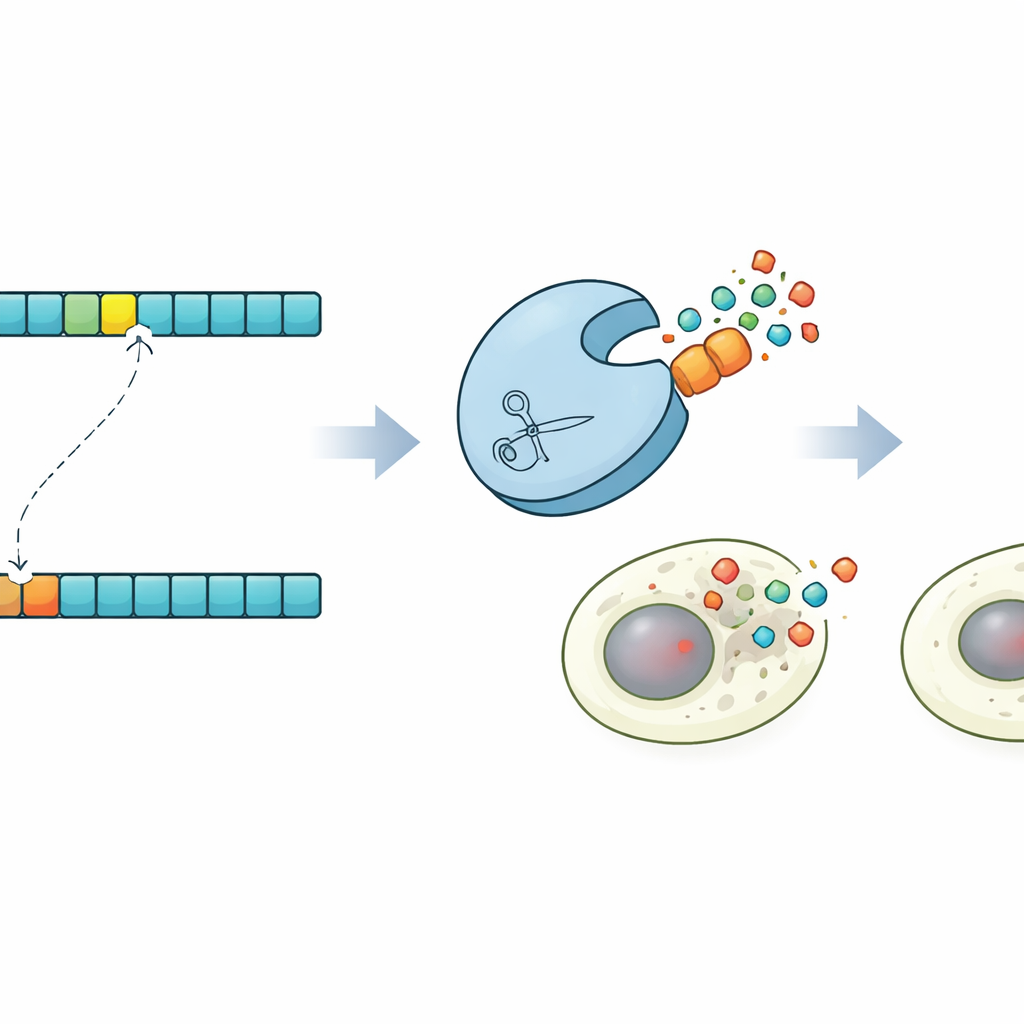
Morte cellulare programmata su comando
Per dimostrare cosa rende possibile questo controllo, i ricercatori hanno progettato una dimostrazione convincente nelle cellule umane. Usando le loro previsioni, hanno identificato una proteasi in grado di tagliare una versione sottilmente mutata di un sito bersaglio lasciando intatta la versione originale. Hanno quindi costruito varianti di Caspase-3, un importante esecutore della morte cellulare programmata, che portano o il sito normale o quello mutato. In popolazioni cellulari miste, la proteasi ingegnerizzata ha attivato selettivamente Caspase-3 solo nelle cellule con la sequenza mutante, innescando la loro apoptosi e risparmiando le cellule vicine. Questo circuito di “sinpoptosi” dimostra che la progettazione di proteasi guidata dalla co-evoluzione può essere sfruttata per rilevare differenze di un singolo aminoacido e convertirle in decisioni di vita o di morte per le cellule.
Cosa significa per il futuro
Per i non specialisti, il messaggio chiave è che gli autori hanno trasformato indizi evolutivi sparsi nelle sequenze proteiche in uno strumento pratico per progettare forbici molecolari. ProSSpeC non solo individua proteasi che superano un punto di riferimento di laboratorio come TEVp, ma spiega anche quali contatti tra enzima e bersaglio contano di più, anche quando agiscono a distanza. Pur con limiti per sequenze molto distanti da quanto trovato in natura, il modello offre già ai ricercatori un modo per sfogliare e riprogettare migliaia di proteasi virali per tagli più netti e specificità personalizzate. A lungo termine, strumenti di questo tipo potrebbero aiutare a costruire terapie cellulari più intelligenti, diagnosi migliori e sistemi programmabili che modificano le reti proteiche nelle cellule con la stessa precisione che CRISPR ha portato sul DNA.
Citazione: Lim Suan, M.B., Ziegler, C., Syed, Z. et al. Identification and engineering of highly functional potyviral proteases in cells using co-evolutionary models. Nat Commun 17, 3257 (2026). https://doi.org/10.1038/s41467-026-69961-5
Parole chiave: ingegneria delle proteasi, modellazione co-evolutiva, biologia sintetica, morte cellulare programmata, specificità delle proteine