Clear Sky Science · pl
Topologicznie ograniczona nieujemna faktoryzacja macierzy dla zmiennych w czasie ekspresji omicznej
Dlaczego śledzenie ukrytych wzorców chorób ma znaczenie
Współczesna medycyna potrafi mierzyć tysiące genów i cząsteczek z jednej próbki krwi lub tkanki. Te obszerne „omiczne” migawki obiecują wcześniejsze diagnozy i bardziej dopasowane terapie, ale są hałaśliwe, wysokowymiarowe i często zbierane jedynie od niewielkiej liczby pacjentów w czasie. W artykule wprowadzono nowe narzędzie matematyczne, zwane TopConNMF, które pomaga przesiać tę złożoność, aby znaleźć stabilne, wiarygodne molekularne sygnały postępu choroby, nawet gdy dane są ograniczone i zmieniają się w ciągu tygodni lub miesięcy.
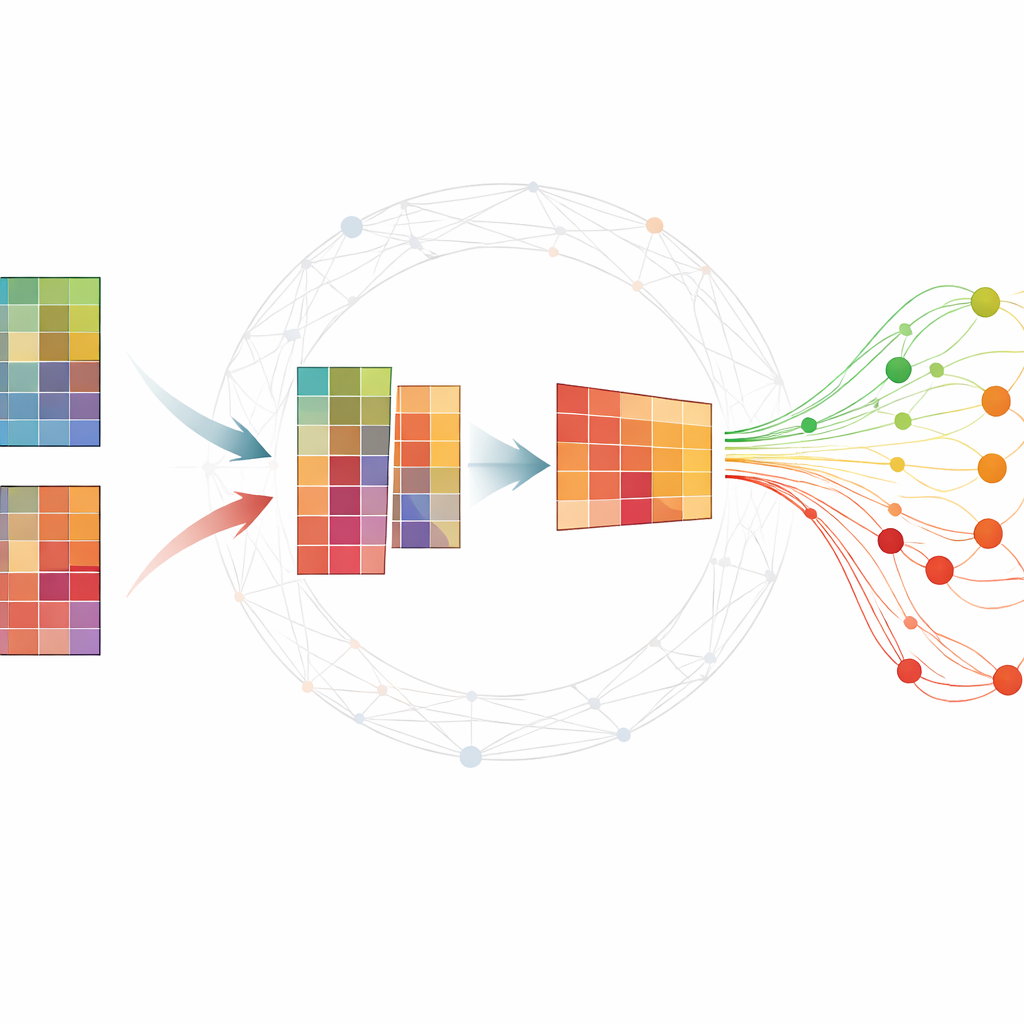
Jak zrozumieć ogromne tabele molekularne
Eksperymenty omiczne zwykle dają olbrzymie tabele, gdzie każdy wiersz to gen lub mała cząsteczka RNA, a każda kolumna to próbka pobrana w określonym czasie. Badacze chcą znaleźć niewielki zestaw cząsteczek — biomarkerów — które podsumowują rozwój choroby i odróżniają osoby chore od zdrowych. Wiele istniejących metod wymaga rozległych, oznaczonych danych, które są trudne do pozyskania, lub zwraca niestabilne wyniki zmieniające się przy powtórnym uruchomieniu analizy. Popularna technika, nieujemna faktoryzacja macierzy (NMF), potrafi skompresować dane do ukrytych wzorców, ale sama w sobie często pomija istotną strukturę biologiczną i jest wrażliwa na szum.
Dodanie wiedzy o sieci do modelu
Autorzy rozszerzają standardowe NMF, wplatając informację o tym, jak geny lub białka współdziałają w sieciach. Ich metoda, TopConNMF, robi dwie rzeczy jednocześnie. Po pierwsze, promuje rozwiązania rzadkie, co oznacza preferowanie zwartego zestawu cech, gdzie tylko podzbiór genów silnie przyczynia się do każdego wzorca. Po drugie, stosuje ograniczenie „topologiczne”, które odzwierciedla, jak blisko powiązane są dwie cząsteczki, nie tylko bezpośrednio, ale także przez wspólnych sąsiadów w sieci. To pomaga algorytmowi traktować geny uczestniczące w tych samych procesach biologicznych jako powiązane, dzięki czemu odkrywane wzorce lepiej odzwierciedlają rzeczywiste ścieżki komórkowe.
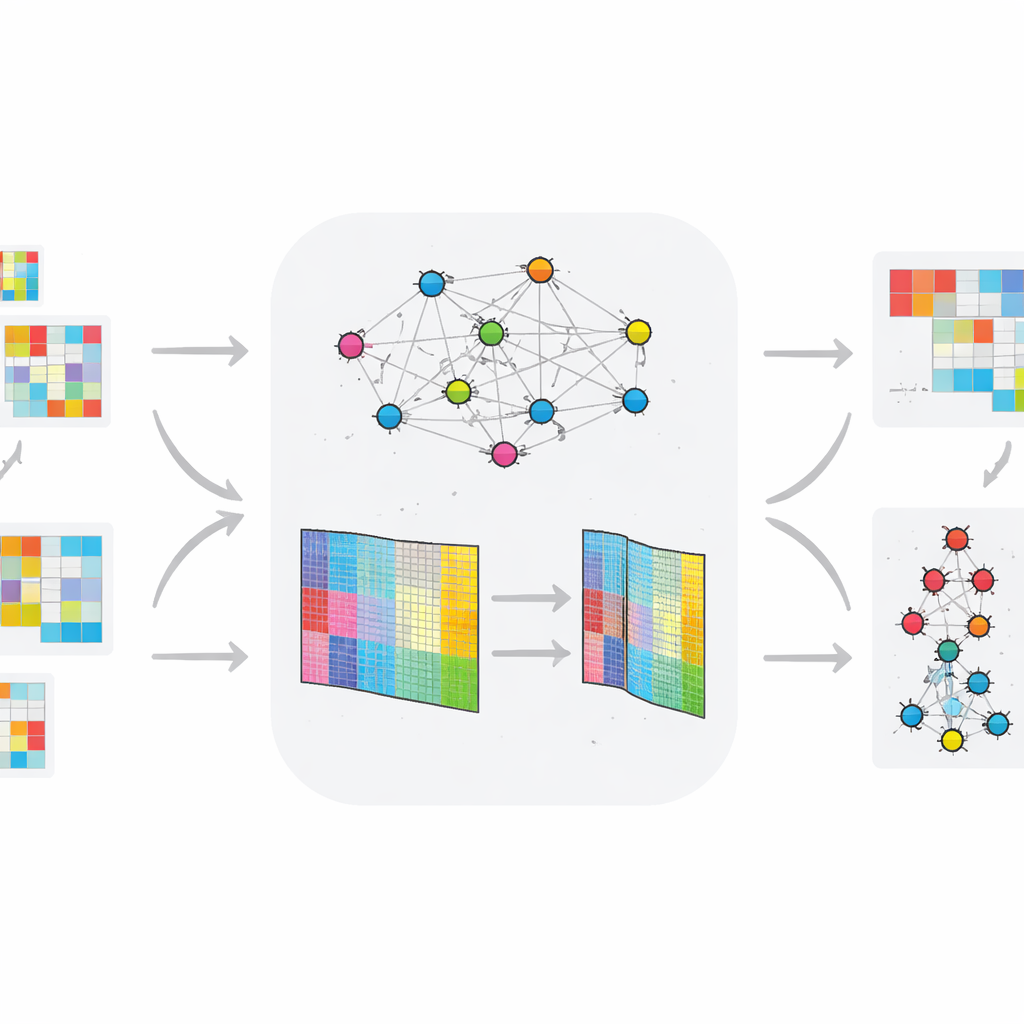
Śledzenie choroby w czasie
W przeciwieństwie do wielu wcześniejszych podejść analizujących dane statyczne, TopConNMF jest zaprojektowany dla omicznych profili zmieniających się w czasie. Autorzy zastosowali metodę do dwóch zestawów danych zwierzęcych: jednego śledzącego aktywność genów u szczurów rozwijających cukrzycę typu 2 na diecie wysokotłuszczowej, oraz drugiego śledzącego małe regulatorowe RNA (miRNA) w modelu choroby Huntingtona. Po skompresowaniu każdego zestawu do mniejszego zbioru wzorców, wyniki trafiają do warstwowego systemu klastrowania, który grupuje cząsteczki na podstawie tego, jak ich zachowanie zmienia się w czasie i między grupami zdrowymi a chorymi. Ten proces wyróżnia cząsteczki, których trajektorie ekspresji najczytelniej rozdzielają zwierzęta narażone od kontrolnych.
Jak dobrze działa nowa metoda
Aby przetestować niezawodność, badacze wielokrotnie uruchamiali TopConNMF z różnymi losowymi punktami startowymi i śledzili, jak dobrze odtwarza on oryginalne dane. Błąd rekonstrukcji stopniowo malał i stabilizował się po około 150 iteracjach, z bardzo małą zmiennością między uruchomieniami, co wskazuje na solidną zbieżność. Porównali też TopConNMF z kilkoma nowoczesnymi metodami na ośmiu benchmarkowych zestawach omicznych, w tym sześciu statycznych i dwóch zmiennych w czasie. W miarach rekonstrukcji danych i jakości klastrowania TopConNMF wypadł co najmniej tak dobrze jak konkurenci, a w wielu przypadkach osiągnął wyższą trafność przy przewidywaniu, które biomarkery rzeczywiście wiążą się z chorobą.
Od wzorców do konkretnych biomarkerów
Co istotne, biomarkery wyeksponowane przez TopConNMF nie są jedynie artefaktami statystycznymi; wiele z nich zgadza się z znaną biologią. W badaniu nad cukrzycą geny często wybierane, takie jak HMGCS2, ACOT1 i PDK4, mają dobrze udokumentowane role w metabolizmie energetycznym, gospodarowaniu tłuszczami i uszkodzeniu serca w cukrzycy. Ich powtarzające się pojawianie się sugeruje, że metoda skutecznie wychwytuje kluczowe zakłócenia metaboliczne, a nie losowy szum. W przypadku choroby Huntingtona zidentyfikowane wzorce miRNA są zgodne z wcześniejszymi pracami łączącymi konkretne małe RNA z uszkodzeniem neuronów i postępem choroby, choć artykuł pozostawia szczegółowe analizy ścieżek wcześniejszym, wyspecjalizowanym badaniom.
Co to oznacza dla przyszłej medycyny
Mówiąc prościej, TopConNMF to inteligentniejszy sposób kompresji ogromnych, czasowych zestawów molekularnych do niewielkiego, biologicznie istotnego zbioru markerów. Szanując sposób, w jaki geny i białka są ze sobą powiązane, oraz faworyzując proste, rzadkie wyjaśnienia, dostarcza stabilne listy biomarkerów z relatywnie niewielu próbek. To może wspierać wcześniejsze diagnozy, lepsze grupowanie pacjentów i bardziej ukierunkowane terapie w złożonych chorobach, takich jak cukrzyca typu 2 czy choroba Huntingtona. W miarę jak technologie omiczne stają się rutyną w klinikach, narzędzia takie jak TopConNMF mogą pomóc zlikwidować przepaść między surowymi danymi molekularnymi a praktycznymi decyzjami medycznymi.
Cytowanie: Dey, A., Sharma, K.D., Chatterjee, A. et al. Topology constrained nonnegative matrix factorization for time varying omic expression. Sci Rep 16, 13285 (2026). https://doi.org/10.1038/s41598-026-43968-w
Słowa kluczowe: odkrywanie biomarkerów, omiczne szeregi czasowe, sieci genowe, faktoryzacja macierzy, postęp choroby