Clear Sky Science · es
Factorización de matrices no negativa con restricción topológica para expresión ómica variable en el tiempo
Por qué importa seguir patrones ocultos de la enfermedad
La medicina moderna puede medir miles de genes y moléculas a partir de una única muestra de sangre o tejido. Estas enormes instantáneas «ómicas» prometen diagnósticos más tempranos y tratamientos más personalizados, pero son ruidosas, de alta dimensión y con frecuencia se recogen de un número reducido de pacientes a lo largo del tiempo. Este artículo presenta una nueva herramienta matemática, llamada TopConNMF, que ayuda a tamizar esta complejidad para encontrar señales moleculares estables y fiables de la progresión de la enfermedad, incluso cuando los datos son limitados y cambian a lo largo de semanas o meses.
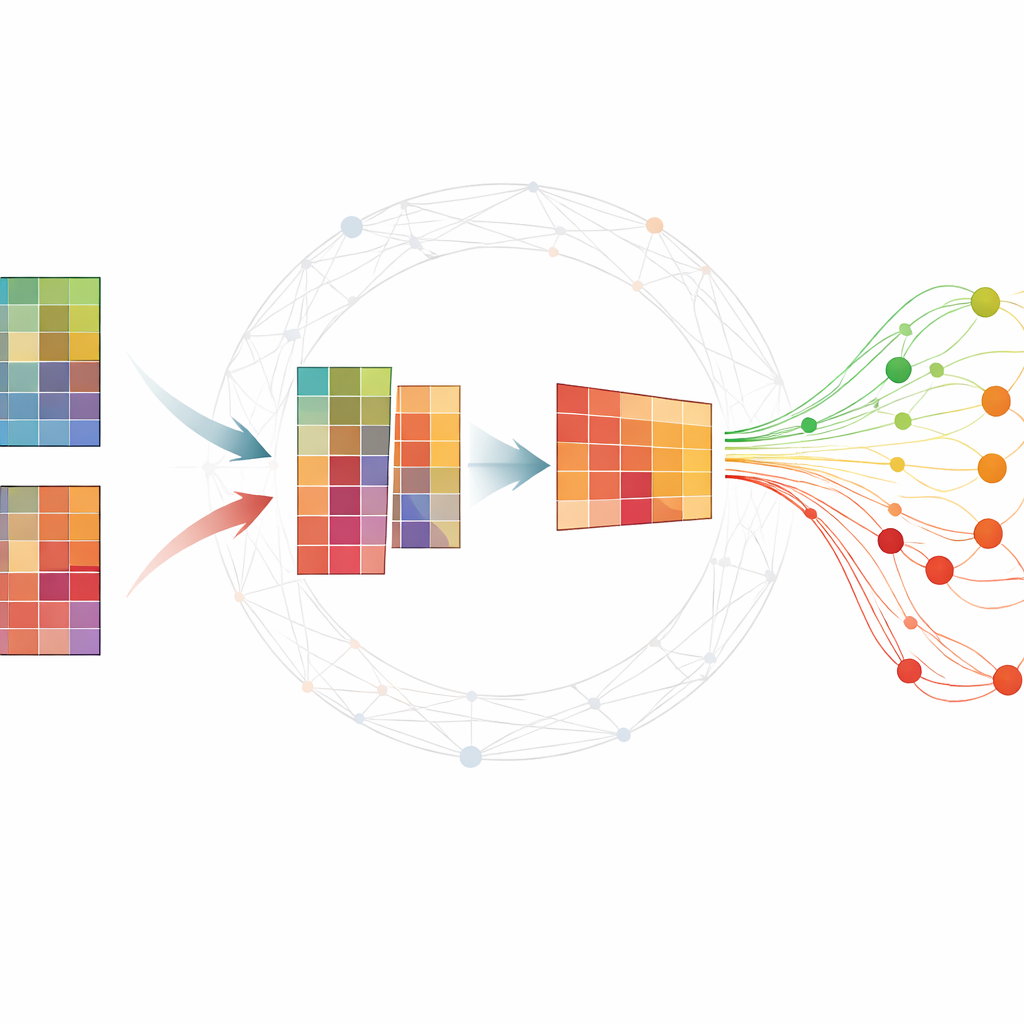
Dar sentido a grandes tablas moleculares
Los experimentos ómicos suelen generar tablas gigantes en las que cada fila es un gen o una pequeña molécula de ARN y cada columna es una muestra tomada en un momento concreto. Los investigadores quieren encontrar un conjunto reducido de moléculas —biomarcadores— que resuman cómo se desarrolla una enfermedad y distingan a sujetos enfermos de sanos. Muchos métodos existentes requieren abundantes datos etiquetados, que son difíciles de obtener, o devuelven resultados inestables que cambian al repetir el análisis. Una técnica popular, la factorización de matrices no negativa (NMF), puede comprimir los datos en patrones subyacentes, pero por sí sola suele pasar por alto estructura biológica importante y puede ser sensible al ruido.
Añadir conocimiento de redes al proceso
Los autores amplían la NMF estándar incorporando información sobre cómo genes o proteínas tienden a funcionar juntos en redes. Su método, TopConNMF, hace dos cosas a la vez. Primero, fomenta soluciones dispersas, es decir, prefiere un conjunto compacto de características en el que solo un subconjunto de genes contribuya fuertemente a cada patrón. Segundo, utiliza una restricción de «topología» que refleja qué tan conectadas están dos moléculas, no solo de forma directa sino también a través de vecinos compartidos en la red. Esto ayuda al algoritmo a tratar como relacionadas a genes que participan en los mismos procesos biológicos, de modo que los patrones que descubre reflejen mejor las vías celulares reales.
Seguir la enfermedad a lo largo del tiempo
A diferencia de muchos enfoques previos que analizan datos estáticos, TopConNMF está diseñado para perfiles ómicos variables en el tiempo. Los autores aplican su método a dos conjuntos de datos animales: uno que sigue la actividad génica en ratas que desarrollan diabetes tipo 2 con una dieta alta en grasas, y otro que rastrea pequeños ARN reguladores (miARN) en un modelo de la enfermedad de Huntington. Tras comprimir cada conjunto de datos en un número menor de patrones, el método introduce los resultados en un sistema de agrupamiento por capas que agrupa moléculas según cómo cambian su comportamiento a lo largo del tiempo y entre grupos sanos y enfermos. Esta tubería destaca las moléculas cuyas trayectorias de expresión separan con mayor claridad a los animales expuestos de los controles.
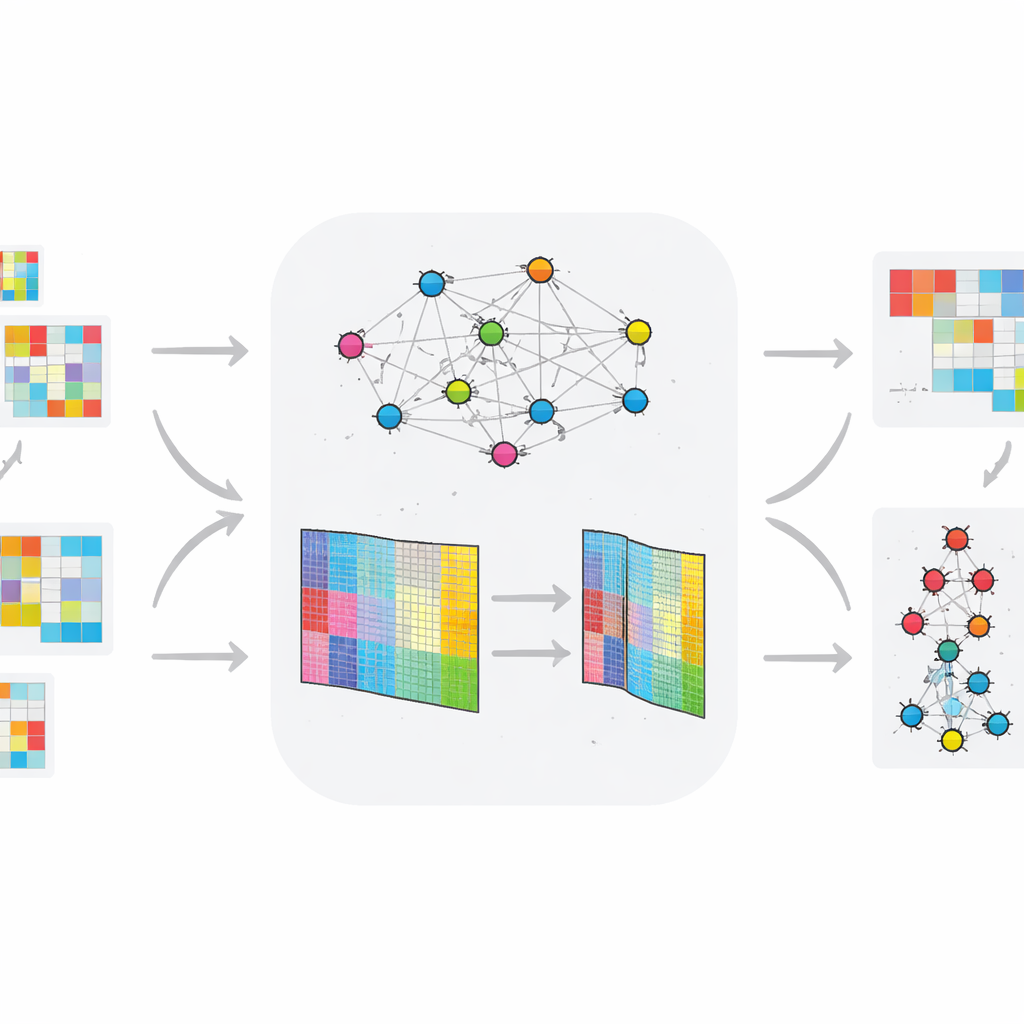
Qué tan bien funciona el nuevo método
Para evaluar la fiabilidad, los investigadores ejecutaron TopConNMF varias veces con distintos puntos de partida aleatorios y observaron cuánto reconstruía los datos originales. El error de reconstrucción disminuyó de forma sostenida y se estabilizó tras unas 150 iteraciones, con muy poca variación entre ejecuciones, lo que indica una convergencia robusta. También compararon TopConNMF con varios métodos de última generación en ocho conjuntos de datos ómicos de referencia, incluidos seis invariables en el tiempo y dos variables. En medidas de reconstrucción de datos y calidad de agrupamiento, TopConNMF rindió igual o mejor que técnicas competidoras y, en muchos casos, produjo mayor precisión al predecir qué biomarcadores se relacionan realmente con la enfermedad.
De los patrones a biomarcadores concretos
De forma crucial, los biomarcadores destacados por TopConNMF no son solo artefactos estadísticos; muchos coinciden con biología conocida. En el estudio de diabetes, genes seleccionados con frecuencia como HMGCS2, ACOT1 y PDK4 tienen roles bien documentados en el metabolismo energético, el manejo de lípidos y el daño cardíaco diabético. Su aparición recurrente sugiere que el método está capturando perturbaciones metabólicas clave y no ruido aleatorio. Para la enfermedad de Huntington, los patrones de miARN identificados son consistentes con trabajos previos que vinculan pequeños ARN específicos con el daño neuronal y la progresión de la enfermedad, aunque el artículo deja el análisis detallado de vías a estudios especializados anteriores.
Qué significa esto para la medicina futura
En términos sencillos, TopConNMF es una forma más inteligente de comprimir enormes conjuntos de datos moleculares basados en el tiempo en un conjunto pequeño y biológicamente significativo de marcadores. Al respetar cómo están conectados genes y proteínas y al favorecer explicaciones simples y dispersas, ofrece listas de biomarcadores estables a partir de relativamente pocas muestras. Esto puede favorecer diagnósticos más tempranos, mejor agrupamiento de pacientes y terapias más dirigidas en enfermedades complejas como la diabetes tipo 2 o la enfermedad de Huntington. A medida que las tecnologías ómicas se vuelvan rutinarias en la clínica, herramientas como TopConNMF podrían ayudar a cerrar la brecha entre los datos moleculares crudos y decisiones médicas accionables.
Cita: Dey, A., Sharma, K.D., Chatterjee, A. et al. Topology constrained nonnegative matrix factorization for time varying omic expression. Sci Rep 16, 13285 (2026). https://doi.org/10.1038/s41598-026-43968-w
Palabras clave: descubrimiento de biomarcadores, ómicas en series temporales, redes génicas, factorización de matrices, progresión de la enfermedad