Clear Sky Science · fr
Factorisation de matrices non négatives contrainte par la topologie pour l’expression omique variant dans le temps
Pourquoi il est important de suivre les motifs cachés de la maladie
La médecine moderne peut désormais mesurer des milliers de gènes et de molécules à partir d’un seul prélèvement sanguin ou tissulaire. Ces vastes instantanés « omiques » promettent un diagnostic plus précoce et des traitements mieux adaptés, mais ils sont bruyants, de très haute dimension, et souvent recueillis sur un petit nombre de patients au cours du temps. Cet article présente un nouvel outil mathématique, appelé TopConNMF, qui aide à trier cette complexité pour identifier des signaux moléculaires stables et fiables de la progression de la maladie, même lorsque les données sont limitées et varient sur des semaines ou des mois.
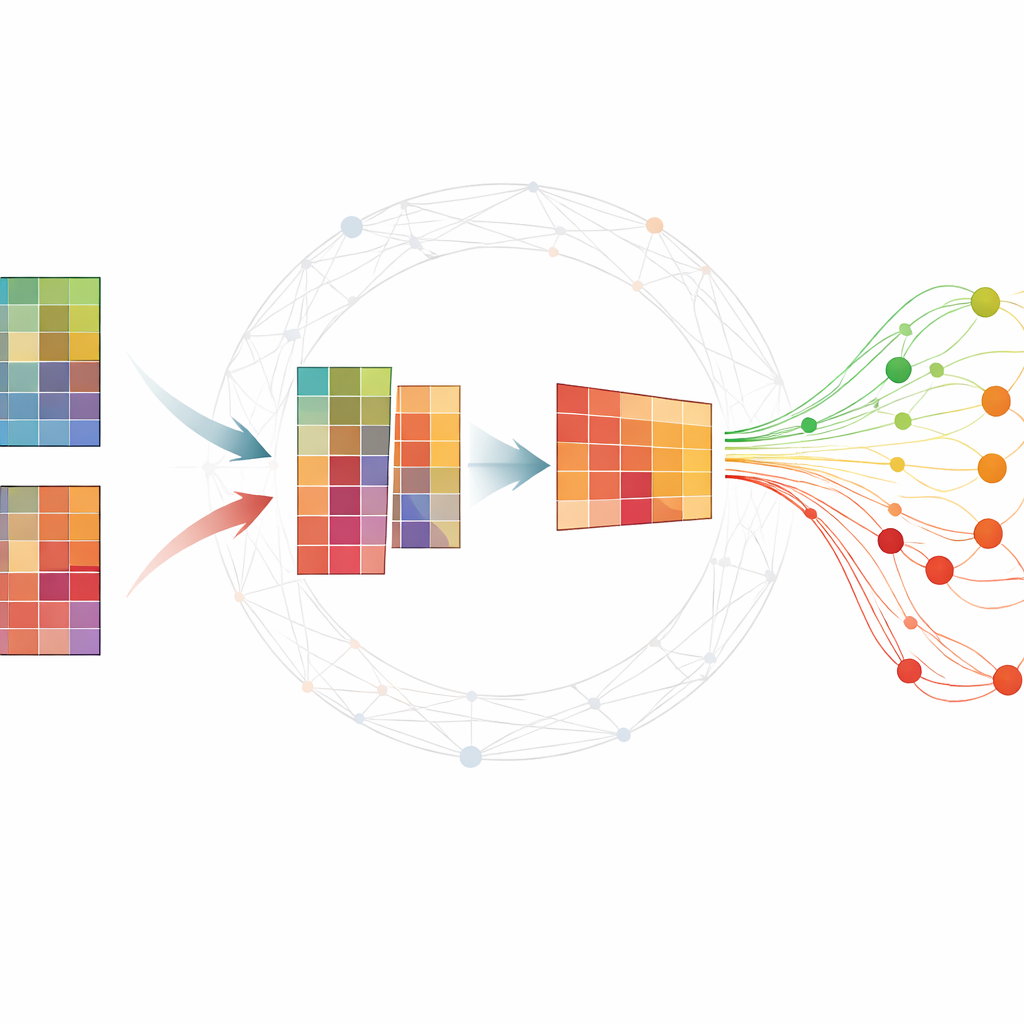
Donner du sens aux grands tableaux moléculaires
Les expériences omiques produisent typiquement d’immenses tableaux où chaque ligne correspond à un gène ou à un petit ARN et chaque colonne à un échantillon prélevé à un moment donné. Les chercheurs cherchent un petit ensemble de molécules — des biomarqueurs — qui résument le développement d’une maladie et distinguent les sujets malades des sujets sains. De nombreuses méthodes existantes nécessitent soit des données étiquetées abondantes, difficiles à obtenir, soit renvoient des résultats instables qui changent à chaque exécution. Une technique populaire, la factorisation de matrices non négatives (NMF), peut compresser les données en motifs sous-jacents, mais prise seule elle passe souvent à côté de structures biologiques importantes et peut être sensible au bruit.
Ajouter la connaissance des réseaux au mélange
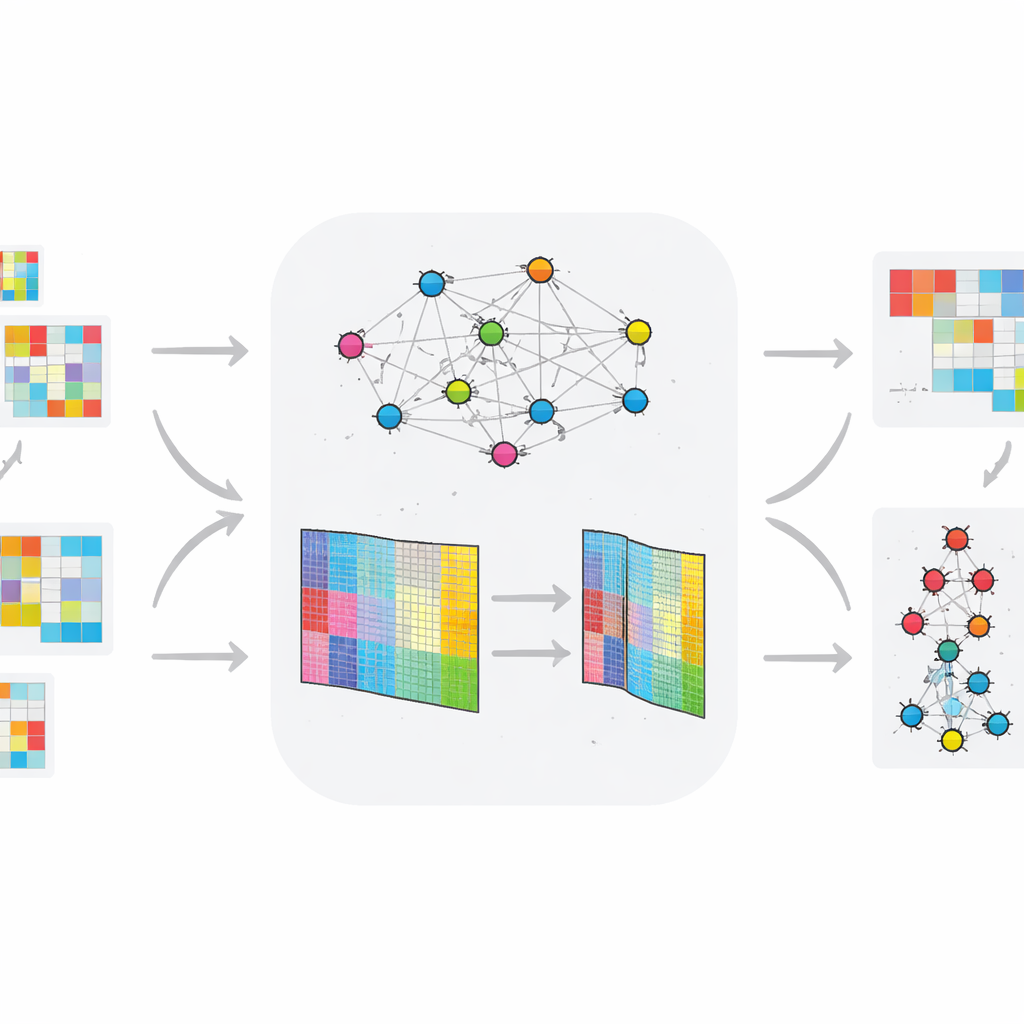
Les auteurs étendent la NMF standard en intégrant des informations sur la façon dont les gènes ou les protéines ont tendance à fonctionner ensemble au sein de réseaux. Leur méthode, TopConNMF, réalise deux objectifs simultanément. D’une part, elle favorise des solutions parcimonieuses, c’est‑à‑dire qu’elle privilégie un ensemble compact de caractéristiques où seule une sous‑partie des gènes contribue fortement à chaque motif. D’autre part, elle utilise une contrainte de « topologie » qui reflète la proximité de deux molécules non seulement directement mais aussi via des voisins communs dans le réseau. Cela aide l’algorithme à considérer comme apparentés des gènes participant aux mêmes processus biologiques, de sorte que les motifs découverts reflètent mieux les voies cellulaires réelles.
Suivre la maladie au fil du temps
Contrairement à de nombreuses approches antérieures qui analysent des données statiques, TopConNMF est conçu pour des profils omiques évoluant dans le temps. Les auteurs appliquent leur méthode à deux jeux de données animaux : l’un suivant l’activité génique chez des rats développant un diabète de type 2 sous régime riche en graisses, et l’autre suivant de petits ARN régulateurs (miARN) dans un modèle de la maladie de Huntington. Après avoir compressé chaque jeu de données en un plus petit nombre de motifs, la méthode alimente les résultats dans un système de clustering en couches qui regroupe les molécules en fonction de l’évolution de leur comportement au fil du temps et entre groupes sains et malades. Ce pipeline met en évidence les molécules dont les trajectoires d’expression séparent le plus clairement les animaux exposés des témoins.
Quelle est la performance de la nouvelle méthode
Pour tester la fiabilité, les chercheurs ont exécuté TopConNMF de manière répétée avec différents points de départ aléatoires et suivi la qualité de la reconstruction des données originales. L’erreur de reconstruction a diminué régulièrement et s’est stabilisée après environ 150 itérations, avec très peu de variation entre les exécutions, indiquant une convergence robuste. Ils ont aussi comparé TopConNMF à plusieurs méthodes de pointe sur huit jeux de données omiques de référence, incluant six collections temporellement invariantes et deux collections variant dans le temps. Selon des mesures de reconstruction des données et de qualité de clustering, TopConNMF a obtenu des performances équivalentes ou supérieures aux techniques concurrentes, et dans de nombreux cas a produit une plus grande précision pour prédire quels biomarqueurs sont réellement liés à la maladie.
Des motifs aux biomarqueurs concrets
De manière cruciale, les biomarqueurs mis en avant par TopConNMF ne sont pas de simples artefacts statistiques ; beaucoup concordent avec la biologie connue. Dans l’étude sur le diabète, des gènes fréquemment sélectionnés tels que HMGCS2, ACOT1 et PDK4 jouent des rôles bien documentés dans le métabolisme énergétique, la gestion des lipides et les lésions cardiaques diabétiques. Leur réapparition suggère que la méthode capture avec succès des perturbations métaboliques clés plutôt que du bruit aléatoire. Pour la maladie de Huntington, les motifs de miARN identifiés sont cohérents avec des travaux antérieurs liant certains petits ARN aux lésions des cellules nerveuses et à la progression de la maladie, bien que l’article renvoie l’analyse détaillée des voies à des études spécialisées antérieures.
Ce que cela signifie pour la médecine de demain
Simplement dit, TopConNMF est une façon plus intelligente de compresser d’immenses jeux de données moléculaires temporels en un petit ensemble de marqueurs biologiquement significatifs. En respectant la manière dont gènes et protéines sont interconnectés et en privilégiant des explications simples et parcimonieuses, il fournit des listes de biomarqueurs stables à partir d’un nombre relativement faible d’échantillons. Cela peut soutenir un diagnostic plus précoce, un meilleur regroupement des patients et des thérapies plus ciblées pour des maladies complexes comme le diabète de type 2 ou la maladie de Huntington. À mesure que les technologies omiques deviendront courantes en clinique, des outils comme TopConNMF pourraient aider à faire le lien entre des données moléculaires brutes et des décisions médicales exploitables.
Citation: Dey, A., Sharma, K.D., Chatterjee, A. et al. Topology constrained nonnegative matrix factorization for time varying omic expression. Sci Rep 16, 13285 (2026). https://doi.org/10.1038/s41598-026-43968-w
Mots-clés: découverte de biomarqueurs