Clear Sky Science · nl
Topologie-geconstrueerde niet-negatieve matrixfactorisatie voor tijdsafhankelijke omische expressie
Waarom het volgen van verborgen ziektepatronen ertoe doet
De moderne geneeskunde kan nu duizenden genen en moleculen meten uit één bloed- of weefselmonster. Deze enorme “omische” momentopnamen beloven vroegere diagnoses en meer op maat gemaakte behandelingen, maar ze zijn ruisachtig, hoogdimensionaal en vaak over slechts een klein aantal patiënten in de tijd verzameld. Dit artikel introduceert een nieuw wiskundig hulpmiddel, TopConNMF, dat helpt deze complexiteit te doorzoeken om stabiele, betrouwbare moleculaire kentekens van ziekteprogressie te vinden, zelfs wanneer gegevens beperkt zijn en in de loop van weken of maanden veranderen.
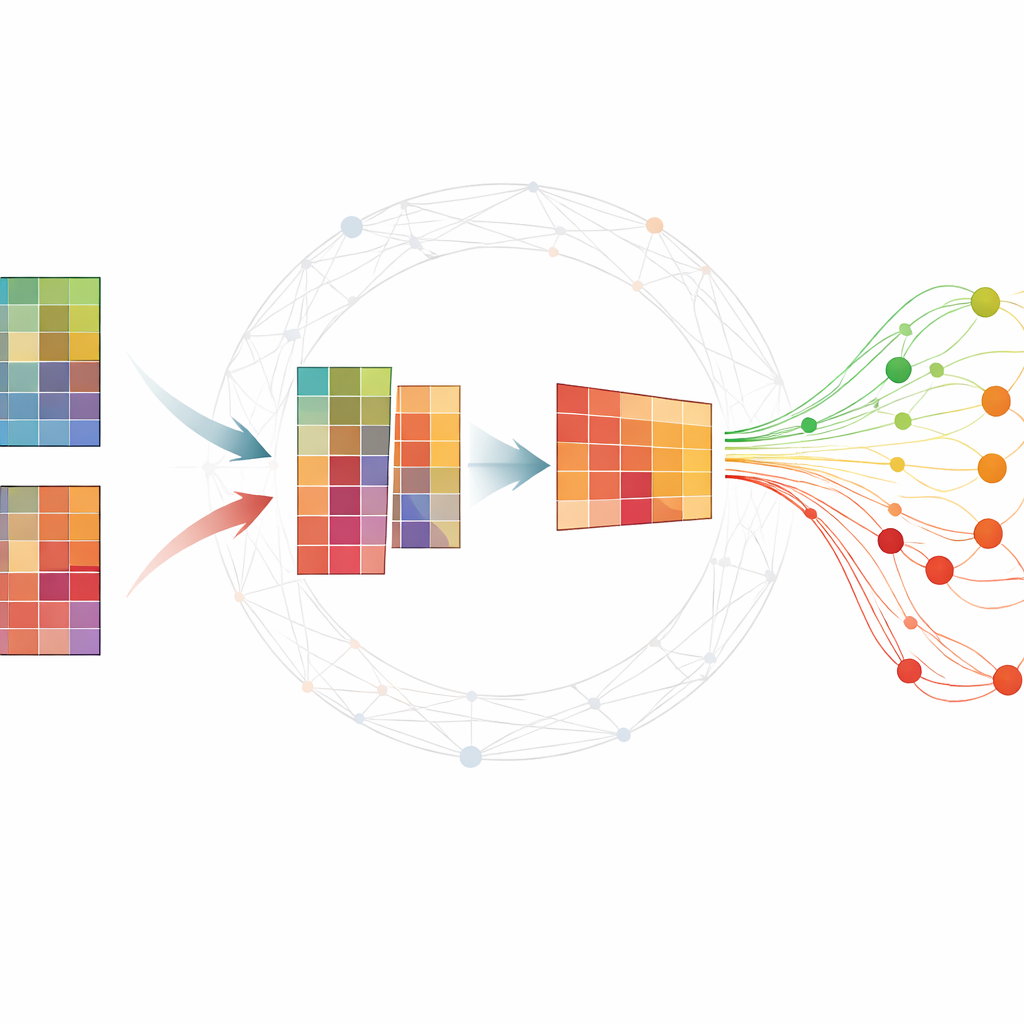
Een grote moleculaire tabel begrijpelijk maken
Omische experimenten produceren doorgaans reusachtige tabellen waarbij elke rij een gen of klein RNA-molecuul is en elke kolom een monster dat op een specifiek tijdstip is genomen. Onderzoekers willen een kleine set moleculen—biomarkers—vinden die samenvatten hoe een ziekte zich ontwikkelt en zieke van gezonde proefpersonen onderscheiden. Veel bestaande methodes hebben ofwel uitgebreide gelabelde data nodig, die moeilijk te verkrijgen zijn, of geven onstabiele resultaten die veranderen bij herhaalde analyses. Een populaire techniek, niet-negatieve matrixfactorisatie (NMF), kan de gegevens comprimeren tot onderliggende patronen, maar op zichzelf mist ze vaak belangrijke biologische structuur en kan ze gevoelig zijn voor ruis.
Netwerkkennis toevoegen
De auteurs breiden standaard NMF uit door informatie te verwerken over hoe genen of eiwitten gewoonlijk samenwerken binnen netwerken. Hun methode, TopConNMF, doet twee dingen tegelijk. Ten eerste stimuleert ze spaarzame oplossingen: ze geeft de voorkeur aan een compacte set kenmerken waarbij slechts een subset genen sterk bijdraagt aan elk patroon. Ten tweede gebruikt ze een “topologie”-beperking die weerspiegelt hoe nauw twee moleculen verbonden zijn, niet alleen direct maar ook via gedeelde buren in het netwerk. Dit helpt het algoritme genen die deelnemen aan dezelfde biologische processen als gerelateerd te behandelen, zodat de gevonden patronen beter echte cellulaire paden weerspiegelen.
De ziekte door de tijd volgen
In tegenstelling tot veel eerdere benaderingen die naar statische gegevens kijken, is TopConNMF ontworpen voor in de tijd variërende omische profielen. De auteurs passen hun methode toe op twee dierlijke datasets: één die genactiviteit volgt bij ratten die type 2-diabetes ontwikkelen onder een vethoudend dieet, en een andere die kleine regulatorische RNA’s (miRNA’s) volgt in een model van de ziekte van Huntington. Nadat elke dataset is teruggebracht tot een kleinere set patronen, voert de methode de resultaten in een gelaagd clusteringssysteem waarmee moleculen worden gegroepeerd op basis van hoe hun gedrag in de tijd verandert en tussen gezonde en zieke groepen. Deze pijplijn benadrukt moleculen waarvan de expressietrajecten het duidelijkst blootgestelde van controledieren scheiden.
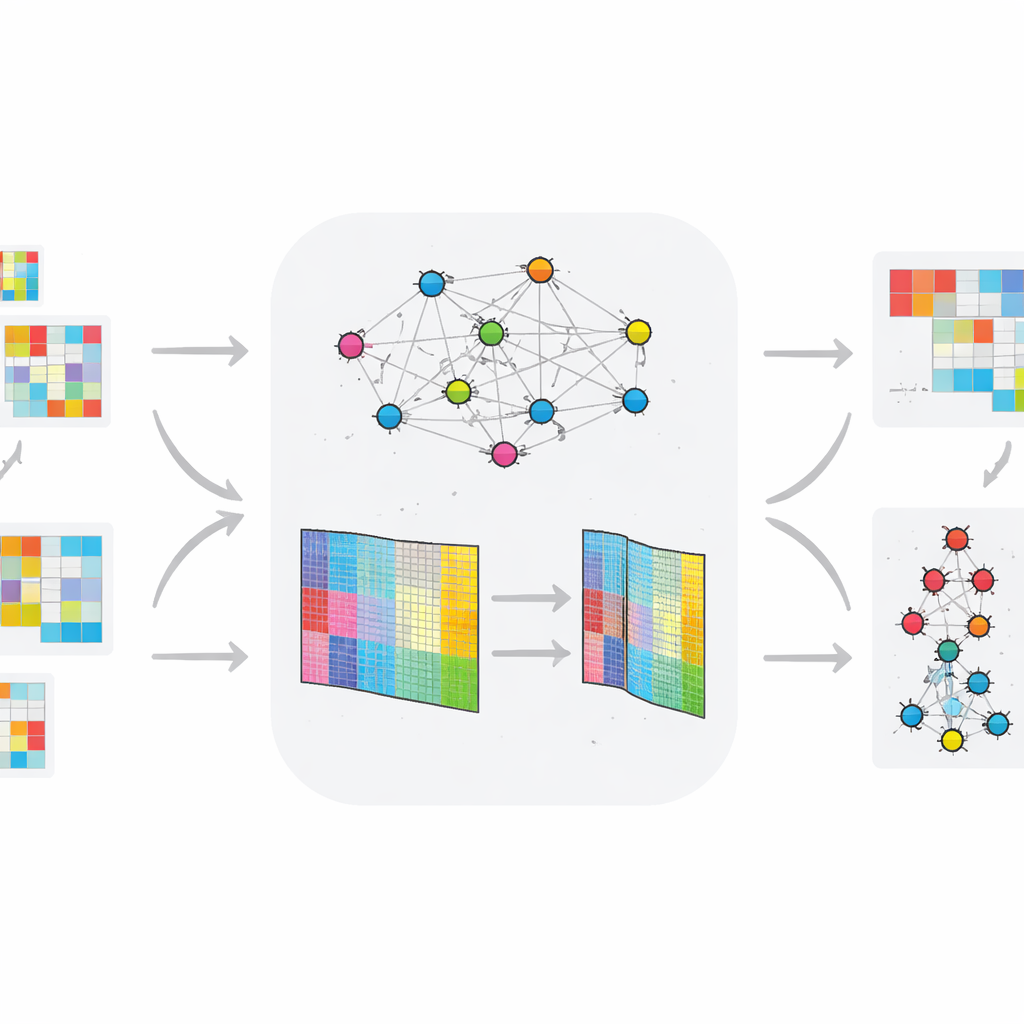
Hoe goed de nieuwe methode presteert
Om de betrouwbaarheid te testen, voerden de onderzoekers TopConNMF herhaaldelijk uit met verschillende willekeurige startpunten en volgden ze hoe goed het de oorspronkelijke gegevens reconstrueerde. De reconstructiefout nam gestaag af en stabiliseerde na ongeveer 150 iteraties, met zeer weinig variatie tussen runs, wat op robuuste convergentie wijst. Ze vergeleken TopConNMF ook met verschillende state-of-the-art methoden op acht benchmark-omische datasets, waaronder zes tijdsinvariante en twee tijdsvariërende verzamelingen. Over maatstaven voor datareconstructie en clusteringkwaliteit presteerde TopConNMF even goed of beter dan concurrerende technieken en leverde in veel gevallen hogere nauwkeurigheid bij het voorspellen welke biomarkers echt met de ziekte verband houden.
Van patronen naar concrete biomarkers
Kritisch is dat de door TopConNMF naar voren gebrachte biomarkers niet slechts statistische artefacten zijn; veel komen overeen met bekende biologie. In de diabetessstudie hebben vaak geselecteerde genen zoals HMGCS2, ACOT1 en PDK4 goed gedocumenteerde rollen in energie metabolisme, vetverwerking en diabetische hartschade. Hun herhaalde verschijning suggereert dat de methode succesvol belangrijke metabole verstoringen vastlegt in plaats van willekeurige ruis. Voor de ziekte van Huntington zijn de geïdentificeerde miRNA-patronen in overeenstemming met eerder werk dat specifieke kleine RNA’s koppelt aan zenuwcelbeschadiging en ziekteprogressie, hoewel het artikel gedetailleerde padanalyses aan eerdere gespecialiseerde studies overlaat.
Wat dit betekent voor de geneeskunde van de toekomst
Simpel gezegd is TopConNMF een slimmere manier om enorme, tijdgebaseerde moleculaire datasets samen te vatten tot een kleine, biologisch zinvolle set markeringen. Door rekening te houden met hoe genen en eiwitten met elkaar verbonden zijn en door te kiezen voor eenvoudige, spaarzame verklaringen, levert het stabiele biomarkerlijsten uit relatief weinig monsters. Dit kan vroegere diagnoses ondersteunen, betere patiëntengroeperingen mogelijk maken en gerichtere therapieën bevorderen bij complexe ziekten zoals type 2-diabetes of de ziekte van Huntington. Naarmate omische technologieën routine worden in klinieken, kunnen tools als TopConNMF helpen de kloof te overbruggen tussen ruwe moleculaire data en bruikbare medische beslissingen.
Bronvermelding: Dey, A., Sharma, K.D., Chatterjee, A. et al. Topology constrained nonnegative matrix factorization for time varying omic expression. Sci Rep 16, 13285 (2026). https://doi.org/10.1038/s41598-026-43968-w
Trefwoorden: biomarkerontdekking, tijdreeks omics, genetwerken, matrixfactorisatie, ziekteprogressie