Clear Sky Science · de
Topologie‑konstruierte nichtnegative Matrixfaktorisierung für zeitlich veränderliche Omics‑Expression
Warum das Verfolgen verborgener Krankheitsmuster wichtig ist
Die moderne Medizin kann inzwischen Tausende von Genen und Molekülen aus einer einzelnen Blut‑ oder Gewebeprobe messen. Diese umfangreichen „Omics“‑Momentaufnahmen versprechen frühere Diagnosen und gezieltere Therapien, sind jedoch rauschbehaftet, hochdimensional und werden oft nur von wenigen Patienten über die Zeit hinweg erhoben. In diesem Artikel wird ein neues mathematisches Werkzeug vorgestellt, TopConNMF, das dabei hilft, diese Komplexität zu durchdringen und stabile, vertrauenswürdige molekulare Wegweiser der Krankheitsprogression zu identifizieren — selbst wenn die Daten begrenzt sind und sich über Wochen oder Monate verändern.
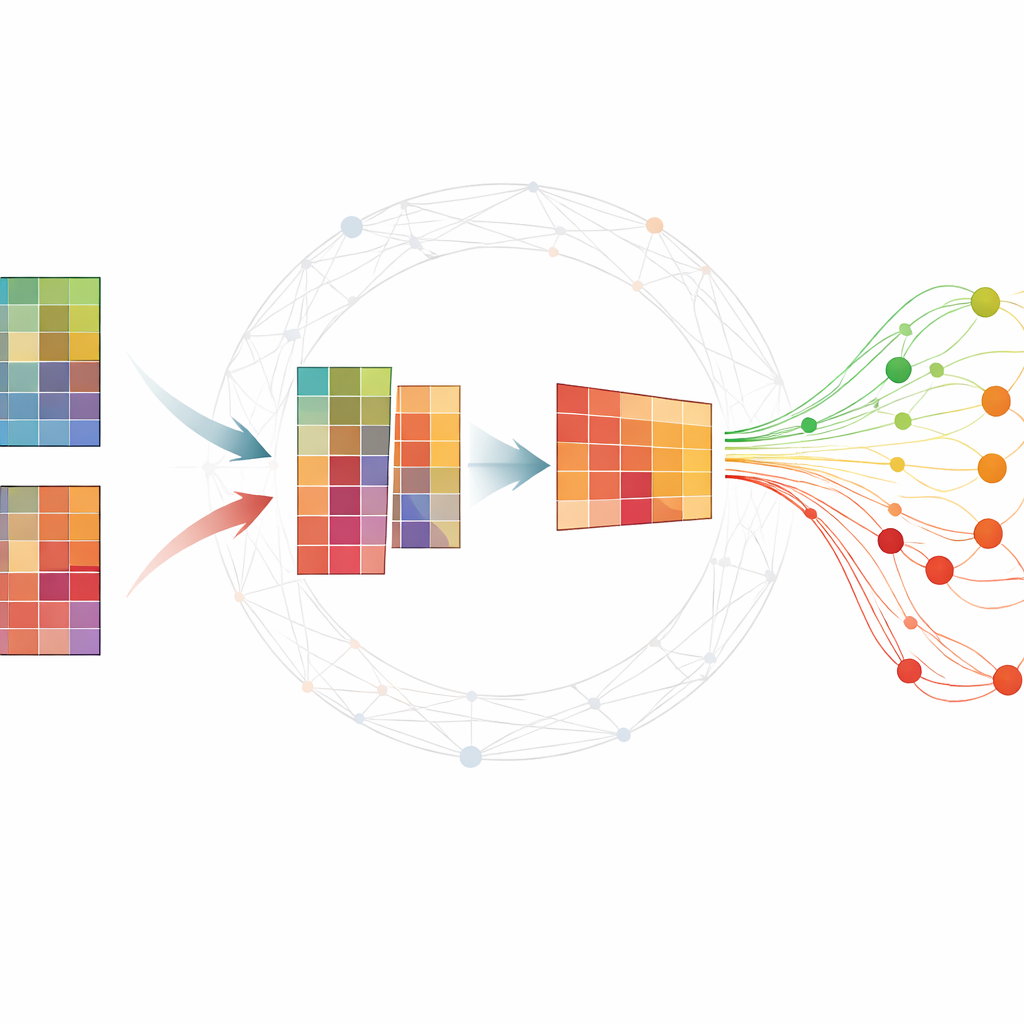
Ordnung in große molekulare Tabellen bringen
Omics‑Experimente erzeugen typischerweise riesige Tabellen, in denen jede Zeile ein Gen oder ein kleines RNA‑Molekül und jede Spalte eine zu einem bestimmten Zeitpunkt entnommene Probe darstellt. Forschende möchten eine kleine Menge von Molekülen — Biomarker — finden, die zusammenfassen, wie sich eine Krankheit entwickelt und Kranke von Gesunden unterscheiden. Viele bestehende Methoden benötigen umfangreich gelabelte Daten, die schwer zu erhalten sind, oder liefern instabile Ergebnisse, die sich bei Wiederholung der Analyse ändern. Eine verbreitete Technik, die nichtnegative Matrixfaktorisierung (NMF), kann die Daten in zugrundeliegende Muster komprimieren, übersieht aber alleinstehend oft wichtige biologische Strukturen und ist anfällig für Rauschen.
Netzwerkwissen in die Analyse einbeziehen
Die Autorinnen und Autoren erweitern die Standard‑NMF, indem sie Informationen darüber einflechten, wie Gene oder Proteine typischerweise in Netzwerken zusammenarbeiten. Ihre Methode, TopConNMF, leistet zwei Dinge zugleich. Erstens begünstigt sie spärliche Lösungen, das heißt sie bevorzugt eine kompakte Menge an Merkmalen, bei denen nur ein Teil der Gene stark zu jedem Muster beiträgt. Zweitens nutzt sie eine „Topologie“‑Beschränkung, die widerspiegelt, wie eng zwei Moleküle miteinander verbunden sind — nicht nur direkt, sondern auch über gemeinsame Nachbarn im Netzwerk. Das hilft dem Algorithmus, Gene, die an denselben biologischen Prozessen beteiligt sind, als verwandt zu behandeln, sodass die entdeckten Muster Zellpfade besser widerspiegeln.
Die Krankheit über die Zeit verfolgen
Im Gegensatz zu vielen früheren Ansätzen, die statische Daten betrachten, ist TopConNMF für zeitlich veränderliche Omics‑Profile ausgelegt. Die Autorinnen und Autoren wenden ihre Methode auf zwei Tierdatensätze an: einen, der die Genaktivität bei Ratten verfolgt, die unter einer fettreichen Ernährung Typ‑2‑Diabetes entwickeln, und einen weiteren, der kleine regulatorische RNAs (miRNAs) in einem Modell der Huntington‑Krankheit untersucht. Nachdem jedes Datenset auf eine kleinere Menge Muster komprimiert wurde, werden die Ergebnisse in ein mehrstufiges Clustering‑System eingespeist, das Moleküle anhand ihres zeitlichen Verhaltens und der Unterschiede zwischen gesunden und erkrankten Gruppen gruppiert. Diese Pipeline hebt Moleküle hervor, deren Expressionsverläufe exponierte und Kontrolltiere am deutlichsten trennen.
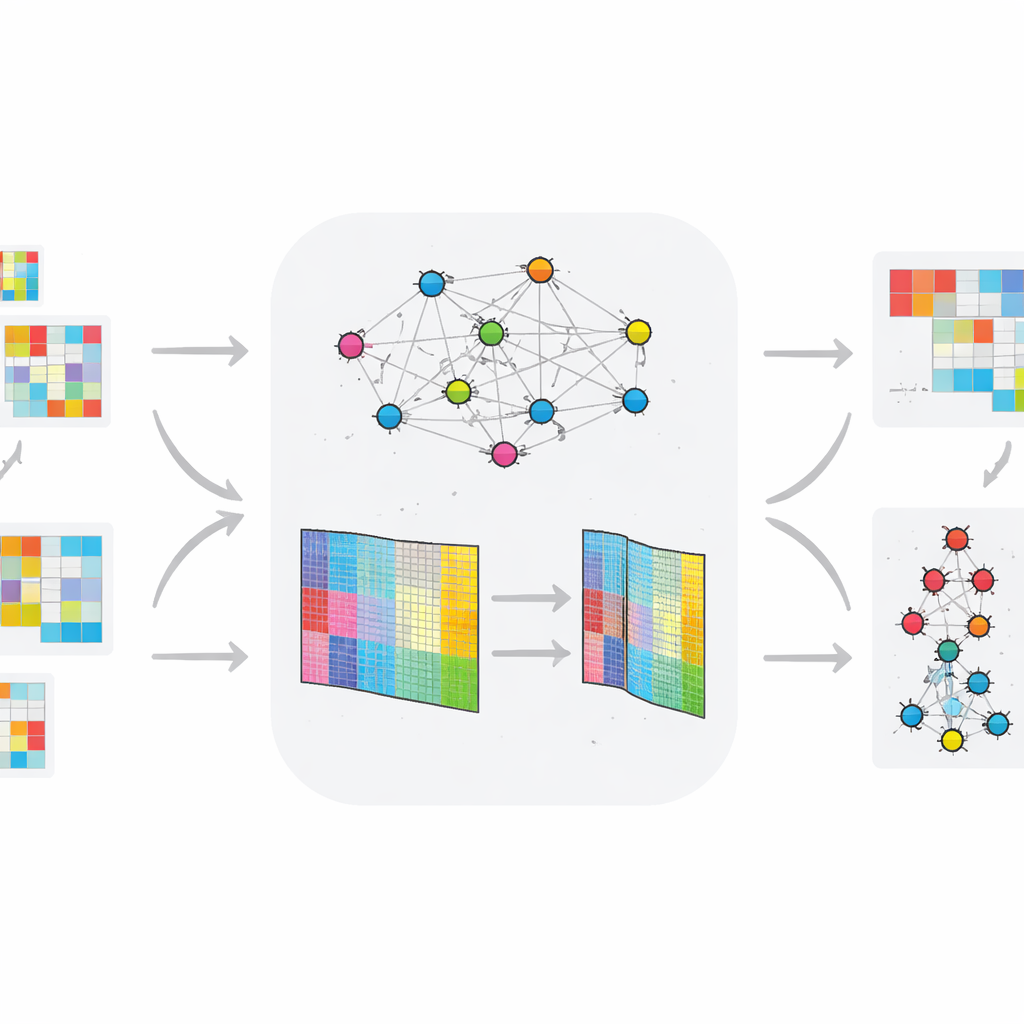
Wie gut die neue Methode arbeitet
Um die Zuverlässigkeit zu testen, führten die Forschenden TopConNMF wiederholt mit verschiedenen zufälligen Startpunkten aus und verfolgten, wie gut die Methode die ursprünglichen Daten rekonstruierte. Der Rekonstruktionsfehler nahm stetig ab und stabilisierte sich nach etwa 150 Iterationen mit sehr geringen Schwankungen zwischen den Ausführungen, was auf eine robuste Konvergenz hindeutet. Sie verglichen TopConNMF außerdem mit mehreren modernen Verfahren an acht Benchmark‑Omics‑Datensätzen, darunter sechs zeitinvariante und zwei zeitveränderliche Sammlungen. Über Messgrößen zur Datenrekonstruktion und Clusterqualität schnitt TopConNMF mindestens genauso gut oder besser ab als konkurrierende Techniken und erreichte in vielen Fällen höhere Genauigkeit bei der Vorhersage, welche Biomarker tatsächlich mit der Krankheit zusammenhängen.
Von Mustern zu konkreten Biomarkern
Wesentlich ist, dass die von TopConNMF hervorgehobenen Biomarker nicht nur statistische Artefakte sind; viele stimmen mit bekannter Biologie überein. In der Diabetes‑Studie haben häufig ausgewählte Gene wie HMGCS2, ACOT1 und PDK4 gut dokumentierte Rollen im Energiestoffwechsel, im Fettstoffwechsel und bei diabetesbedingten Herzschäden. Ihr wiederholtes Auftreten legt nahe, dass die Methode wichtige metabolische Störungen und nicht zufälliges Rauschen erfasst. Für die Huntington‑Krankheit sind die identifizierten miRNA‑Muster mit früheren Arbeiten vereinbar, die bestimmte kleine RNAs mit Nervenzellschäden und Krankheitsprogression verknüpfen, wobei die detaillierte Pfadanalyse spezialisierten Vorstudien überlassen bleibt.
Was das für die Medizin der Zukunft bedeutet
Kurz gesagt ist TopConNMF ein klügerer Weg, riesige, zeitbasierte molekulare Datensätze auf eine kleine, biologisch sinnvolle Menge von Markern zu verdichten. Indem es respektiert, wie Gene und Proteine miteinander vernetzt sind, und indem es einfache, spärliche Erklärungen bevorzugt, liefert es stabile Biomarker‑Listen aus vergleichsweise wenigen Proben. Das kann frühere Diagnosen, bessere Patientengruppierungen und gezieltere Therapien bei komplexen Erkrankungen wie Typ‑2‑Diabetes oder Huntington‑Krankheit unterstützen. Wenn Omics‑Technologien in Kliniken Routine werden, könnten Werkzeuge wie TopConNMF helfen, die Lücke zwischen rohen molekularen Daten und verwertbaren medizinischen Entscheidungen zu schließen.
Zitation: Dey, A., Sharma, K.D., Chatterjee, A. et al. Topology constrained nonnegative matrix factorization for time varying omic expression. Sci Rep 16, 13285 (2026). https://doi.org/10.1038/s41598-026-43968-w
Schlüsselwörter: Biomarker‑Entdeckung, zeitliche Omics, Gen‑Netzwerke, Matrixfaktorisierung, Krankheitsprogression