Clear Sky Science · en
Topology constrained nonnegative matrix factorization for time varying omic expression
Why tracking hidden disease patterns matters
Modern medicine can now measure thousands of genes and molecules from a single blood or tissue sample. These vast “omic” snapshots promise earlier diagnosis and more tailored treatments, but they are noisy, high‑dimensional, and often collected from only a small number of patients over time. This paper introduces a new mathematical tool, called TopConNMF, that helps sift through this complexity to find stable, trustworthy molecular signposts of disease progression, even when data are limited and change across weeks or months.
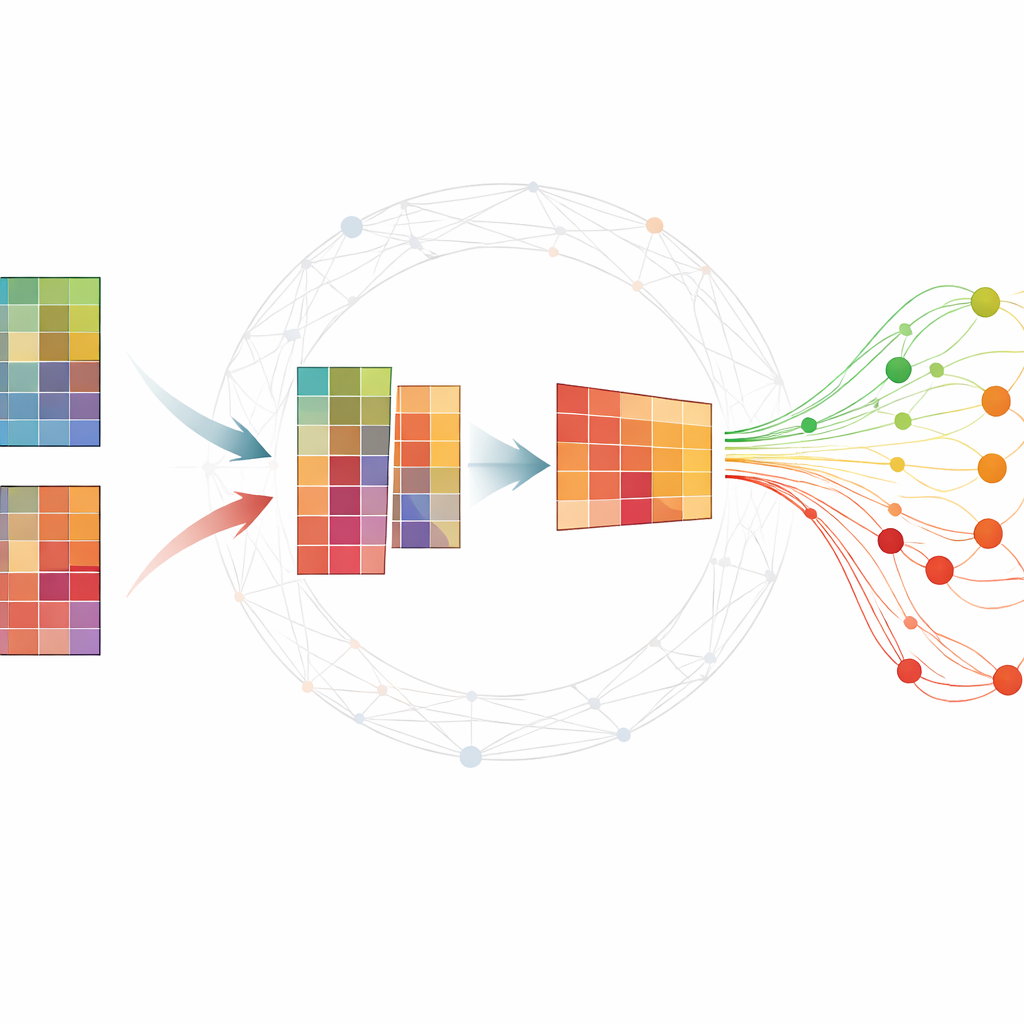
Making sense of big molecular tables
Omic experiments typically produce giant tables where each row is a gene or small RNA molecule and each column is a sample taken at a specific time. Researchers want to find a small set of molecules—biomarkers—that summarize how a disease develops and distinguish sick from healthy subjects. Many existing methods either need extensive labeled data, which are hard to obtain, or return unstable results that change when the analysis is rerun. A popular technique, nonnegative matrix factorization (NMF), can compress the data into underlying patterns, but by itself it often misses important biological structure and can be sensitive to noise.
Adding network knowledge to the mix
The authors extend standard NMF by weaving in information about how genes or proteins tend to work together in networks. Their method, TopConNMF, does two things at once. First, it encourages sparse solutions, meaning it prefers a compact set of features where only a subset of genes strongly contribute to each pattern. Second, it uses a "topology" constraint that reflects how closely connected any two molecules are, not just directly but also through shared neighbors in the network. This helps the algorithm treat genes that participate in the same biological processes as related, so the patterns it uncovers better mirror real cellular pathways.
Following disease over time
Unlike many earlier approaches that look at static data, TopConNMF is designed for time‑varying omic profiles. The authors apply their method to two animal datasets: one tracking gene activity in rats developing type 2 diabetes under a high‑fat diet, and another tracking small regulatory RNAs (miRNAs) in a model of Huntington’s disease. After compressing each dataset into a smaller set of patterns, the method feeds the results into a layered clustering system that groups molecules based on how their behavior changes across time and between healthy and diseased groups. This pipeline highlights molecules whose expression trajectories most clearly separate exposed from control animals.
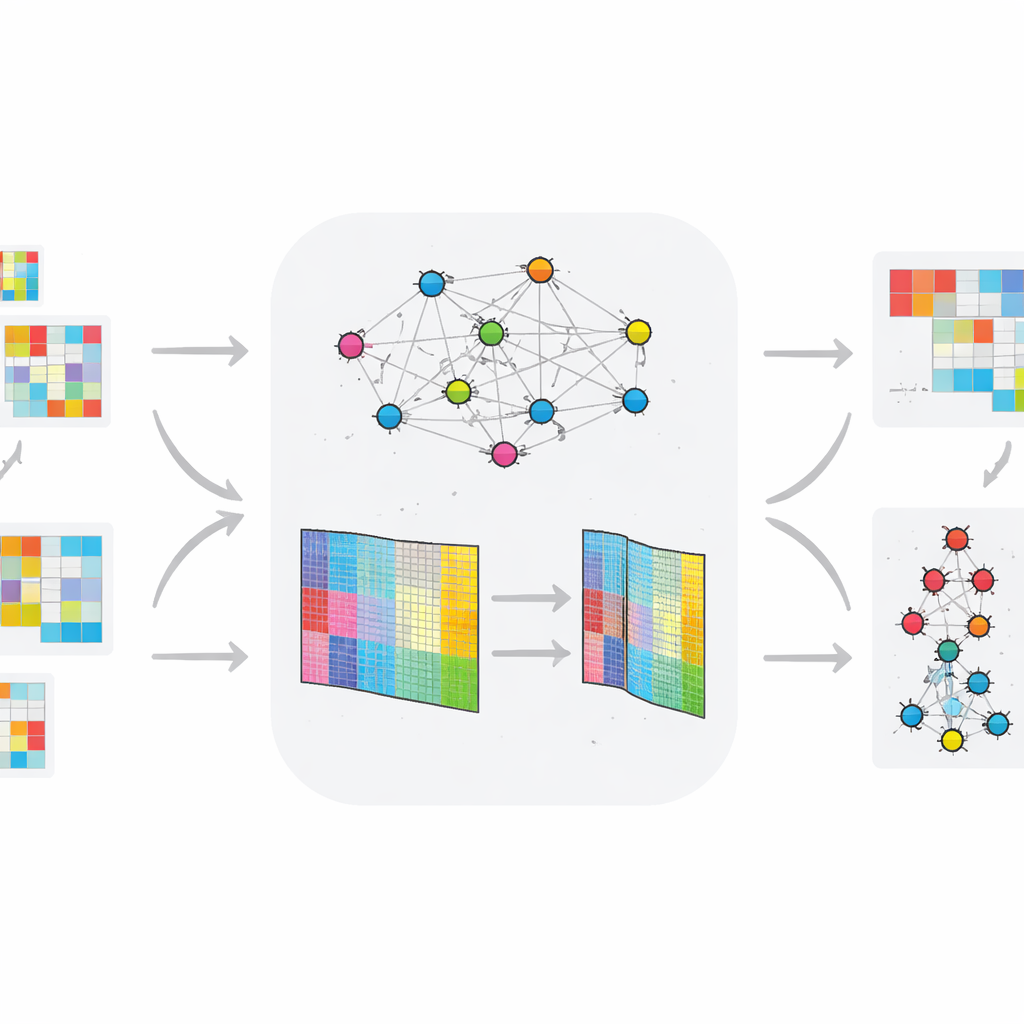
How well the new method performs
To test reliability, the researchers repeatedly ran TopConNMF with different random starting points and tracked how well it rebuilt the original data. The reconstruction error steadily decreased and stabilized after about 150 iterations, with very little variation between runs, indicating robust convergence. They also compared TopConNMF to several state‑of‑the‑art methods on eight benchmark omic datasets, including six time‑invariant and two time‑varying collections. Across measures of data reconstruction and clustering quality, TopConNMF performed as well as or better than competing techniques, and in many cases produced higher accuracy when predicting which biomarkers truly relate to disease.
From patterns to concrete biomarkers
Crucially, the biomarkers highlighted by TopConNMF are not just statistical artifacts; many align with known biology. In the diabetes study, frequently selected genes such as HMGCS2, ACOT1, and PDK4 have well‑documented roles in energy metabolism, fat handling, and diabetic heart damage. Their repeated appearance suggests that the method is successfully capturing key metabolic disruptions rather than random noise. For Huntington’s disease, the identified miRNA patterns are consistent with previous work linking specific small RNAs to nerve cell damage and disease progression, although the paper leaves detailed pathway analysis to earlier specialized studies.
What this means for future medicine
In plain terms, TopConNMF is a smarter way to compress huge, time‑based molecular datasets into a small, biologically meaningful set of markers. By respecting how genes and proteins are wired together and by favoring simple, sparse explanations, it delivers stable biomarker lists from relatively few samples. This can support earlier diagnosis, better grouping of patients, and more targeted therapies in complex diseases such as type 2 diabetes or Huntington’s disease. As omic technologies become routine in clinics, tools like TopConNMF could help bridge the gap between raw molecular data and actionable medical decisions.
Citation: Dey, A., Sharma, K.D., Chatterjee, A. et al. Topology constrained nonnegative matrix factorization for time varying omic expression. Sci Rep 16, 13285 (2026). https://doi.org/10.1038/s41598-026-43968-w
Keywords: biomarker discovery, time series omics, gene networks, matrix factorization, disease progression