Clear Sky Science · it
Fattorizzazione matriciale non negativa vincolata dalla topologia per espressione omica che varia nel tempo
Perché è importante seguire i modelli nascosti delle malattie
La medicina moderna può ora misurare migliaia di geni e molecole da un singolo campione di sangue o tessuto. Questi vasti “snapshot” omici promettono diagnosi più precoci e terapie più personalizzate, ma sono rumorosi, ad alta dimensionalità e spesso raccolti solo su un numero limitato di pazienti nel tempo. Questo articolo presenta un nuovo strumento matematico, chiamato TopConNMF, che aiuta a setacciare questa complessità per trovare segnali molecolari stabili e affidabili della progressione della malattia, anche quando i dati sono scarsi e cambiano nel corso di settimane o mesi.
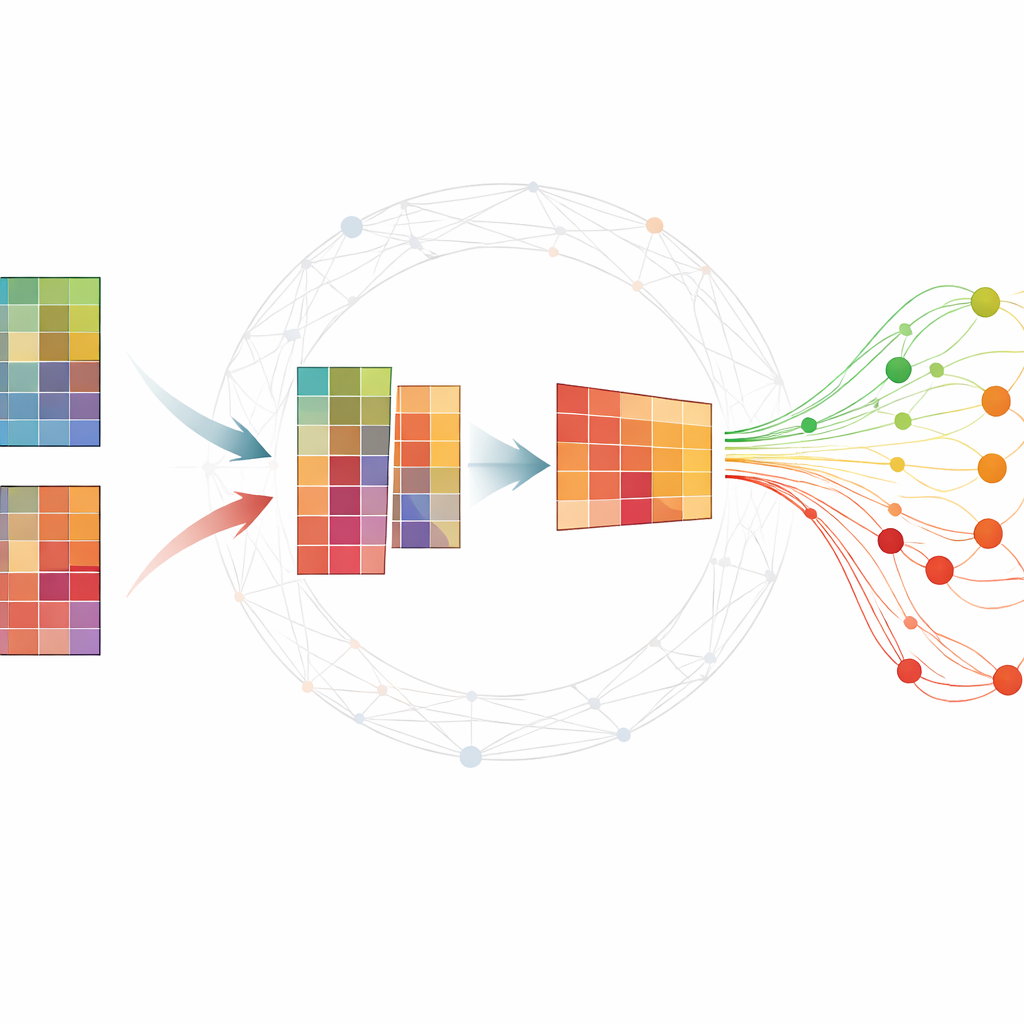
Dare senso a grandi tabelle molecolari
Gli esperimenti omici tipicamente producono tabelle gigantesche in cui ogni riga corrisponde a un gene o a una piccola molecola di RNA e ogni colonna è un campione prelevato in un tempo specifico. I ricercatori vogliono trovare un piccolo insieme di molecole—biomarcatori—that riassumano come si sviluppa una malattia e distinguano soggetti malati da sani. Molti metodi esistenti richiedono grandi quantità di dati etichettati, difficili da ottenere, o restituiscono risultati instabili che cambiano a ogni esecuzione. Una tecnica popolare, la fattorizzazione matriciale non negativa (NMF), può comprimere i dati in modelli sottostanti, ma da sola spesso perde importanti strutture biologiche ed è sensibile al rumore.
Aggiungere la conoscenza delle reti
Gli autori estendono la NMF standard intrecciando informazioni su come geni o proteine tendono a lavorare insieme nelle reti. Il loro metodo, TopConNMF, fa due cose contemporaneamente. Primo, favorisce soluzioni sparse, cioè predilige un insieme compatto di caratteristiche in cui solo un sottoinsieme di geni contribuisce fortemente a ogni modello. Secondo, utilizza un vincolo di “topologia” che riflette quanto due molecole siano strettamente connesse, non solo direttamente ma anche attraverso vicini condivisi nella rete. Questo aiuta l’algoritmo a trattare come correlate le vie cellulari in cui partecipano gli stessi geni, così i modelli scoperti rispecchiano meglio i reali processi biologici.
Seguire la malattia nel tempo
A differenza di molti approcci precedenti che analizzano dati statici, TopConNMF è progettato per profili omici che variano nel tempo. Gli autori applicano il loro metodo a due set di dati animali: uno che traccia l’attività genica in ratti che sviluppano diabete di tipo 2 con una dieta ad alto contenuto di grassi, e un altro che monitora piccoli RNA regolatori (miRNA) in un modello di malattia di Huntington. Dopo aver compresso ciascun dataset in un insieme ridotto di modelli, il metodo inserisce i risultati in un sistema di clustering stratificato che raggruppa le molecole in base a come il loro comportamento cambia nel tempo e tra gruppi sani e malati. Questa pipeline mette in evidenza le molecole le cui traiettorie di espressione separano più nettamente gli animali esposti dai controlli.
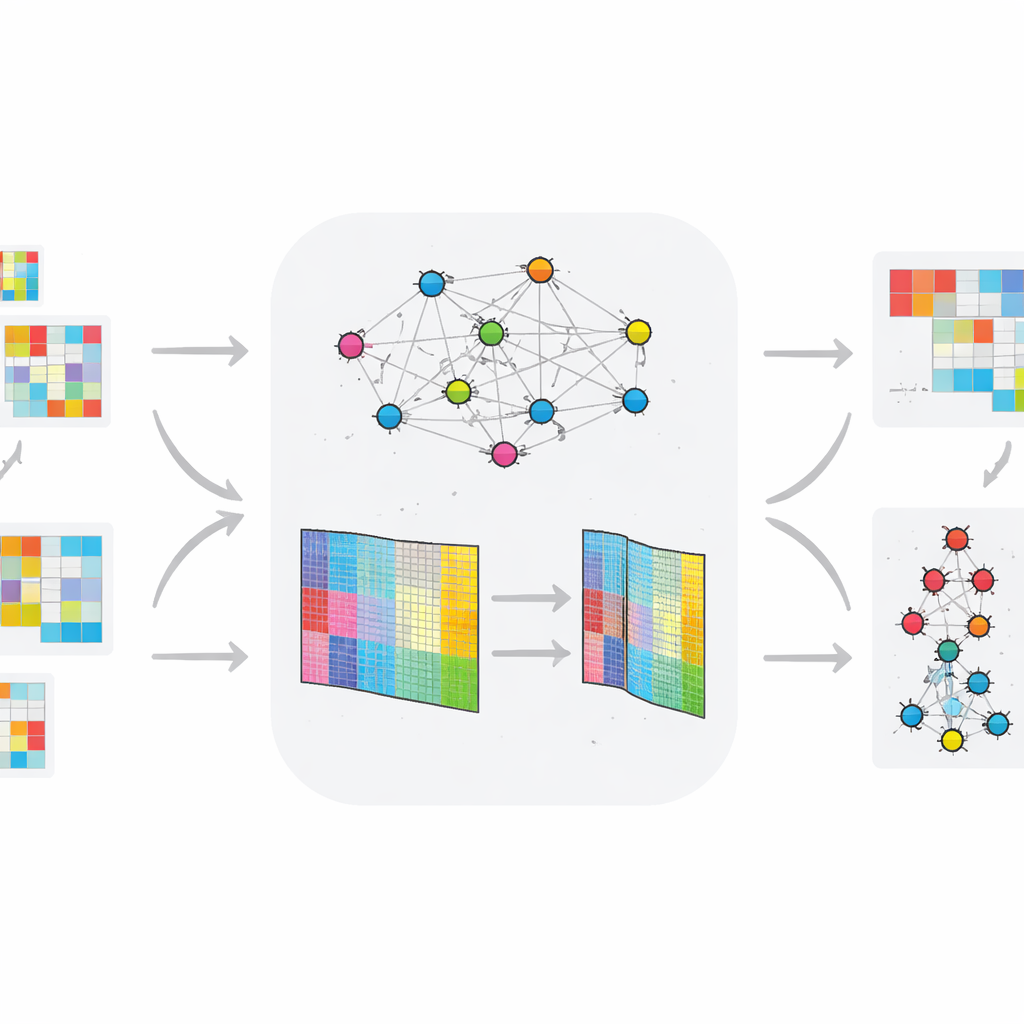
Quanto bene funziona il nuovo metodo
Per testarne l’affidabilità, i ricercatori hanno eseguito ripetutamente TopConNMF con diversi punti di partenza casuali e monitorato quanto bene ricostruiva i dati originali. L’errore di ricostruzione è diminuito in modo costante e si è stabilizzato dopo circa 150 iterazioni, con pochissima variazione tra le esecuzioni, indicando una convergenza robusta. Hanno anche confrontato TopConNMF con diversi metodi all’avanguardia su otto dataset omici benchmark, inclusi sei collezioni tempo-invarianti e due variabili nel tempo. Sulle misure di ricostruzione dei dati e qualità del clustering, TopConNMF ha ottenuto prestazioni pari o superiori alle tecniche concorrenti, e in molti casi ha prodotto maggiore accuratezza nella previsione di quali biomarcatori sono veramente associati alla malattia.
Dai modelli a biomarcatori concreti
Fondamentalmente, i biomarcatori evidenziati da TopConNMF non sono solo artefatti statistici; molti coincidono con conoscenze biologiche consolidate. Nello studio sul diabete, geni frequentemente selezionati come HMGCS2, ACOT1 e PDK4 hanno ruoli ben documentati nel metabolismo energetico, nella gestione dei lipidi e nel danno cardiaco diabetico. La loro ricomparsa suggerisce che il metodo cattura effettivamente perturbazioni metaboliche chiave piuttosto che rumore casuale. Per la malattia di Huntington, i pattern di miRNA identificati sono coerenti con lavori precedenti che collegano specifici piccoli RNA al danno dei neuroni e alla progressione della malattia, sebbene l’articolo rimandi ad analisi di pathway dettagliate svolte in studi specialistici precedenti.
Cosa significa per la medicina del futuro
In termini semplici, TopConNMF è un modo più intelligente di comprimere enormi dataset molecolari temporali in un piccolo insieme di marcatori biologicamente significativi. Rispettando come geni e proteine sono interconnessi e privilegiando spiegazioni semplici e sparse, fornisce elenchi di biomarcatori stabili a partire da un numero relativamente ridotto di campioni. Questo può favorire diagnosi più precoci, una migliore stratificazione dei pazienti e terapie più mirate in malattie complesse come il diabete di tipo 2 o la malattia di Huntington. Con il diffondersi delle tecnologie omiche in ambito clinico, strumenti come TopConNMF potrebbero contribuire a colmare il divario tra dati molecolari grezzi e decisioni mediche azionabili.
Citazione: Dey, A., Sharma, K.D., Chatterjee, A. et al. Topology constrained nonnegative matrix factorization for time varying omic expression. Sci Rep 16, 13285 (2026). https://doi.org/10.1038/s41598-026-43968-w
Parole chiave: scoperta di biomarcatori, omiche a serie temporali, reti geniche, fattorizzazione di matrici, progressione della malattia