Clear Sky Science · it
Indagine integrata DFT, docking molecolare e dinamica molecolare di alcuni nuovi analoghi di 2-tioidantoina come potenti inibitori di CDK2 per la terapia antitumorale
Perché questa ricerca è importante per il trattamento futuro del cancro
I farmaci antitumorali spesso agiscono come strumenti poco selettivi, danneggiando le cellule sane mentre attaccano i tumori. Questo articolo esplora una strategia più precisa: progettare piccole molecole che spengano selettivamente un motore chiave della divisione cellulare, una proteina chiamata CDK2. Utilizzando simulazioni computazionali avanzate invece del metodo empirico della chimica da prove ed errori, i ricercatori identificano diversi candidati promettenti che potrebbero un giorno diventare medicinali antitumorali più sicuri e mirati.
Fermare la divisione cellulare incontrollata alla sua origine
Molti tumori crescono perché il loro ciclo cellulare—l’orologio interno che controlla quando le cellule si dividono—è fuori controllo. CDK2 è uno degli interruttori principali che spingono le cellule attraverso questo ciclo. I farmaci precedenti che cercavano di bloccare CDK2 hanno mostrato potenzialità, ma spesso colpivano anche proteine correlate essenziali per le cellule normali, causando effetti collaterali. Gli autori si concentrano su una famiglia chimica poco esplorata chiamata 2‑tioidantoine, molecole ad anello che possono essere modulate elettronicamente e strutturalmente. L’obiettivo è valutare se membri selezionati di questa famiglia possano legarsi a CDK2 più fortemente, e con maggiore selettività, rispetto alla molecola energetica naturale della cellula, l’ATP, evitando nel contempo altre chinasi.
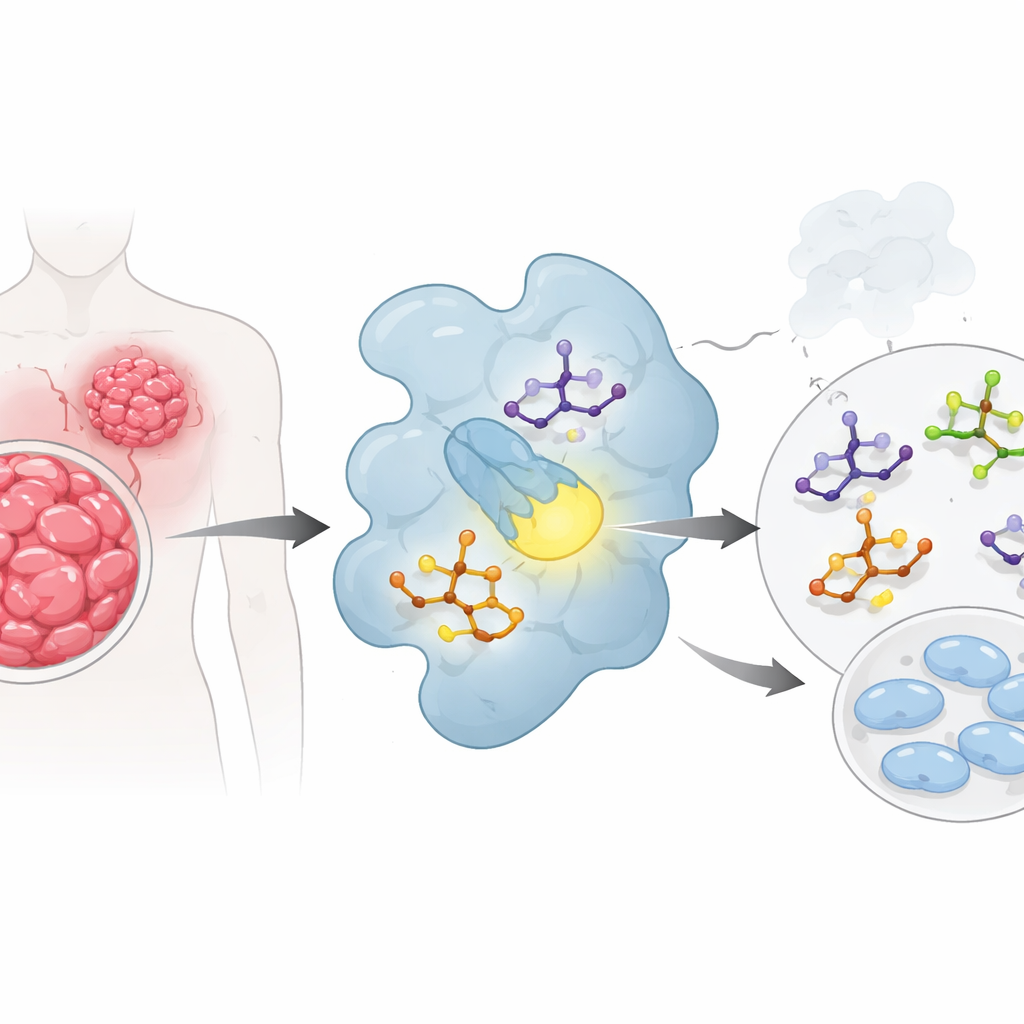
Usare i computer per “pre-testare” i candidati farmaci
Invece di andare direttamente al banco di laboratorio, il team ha prima impiegato diversi livelli di chimica quantistica e modellazione molecolare per capire come si comportano le loro molecole. Hanno calcolato quanto facilmente ogni composto può spostare gli elettroni nella sua struttura, quanto è polare e quali parti della molecola sono più propense a formare contatti favorevoli con una proteina. I composti etichettati 2b fino a 2e si sono distinti: presentavano piccoli gap energetici tra i loro stati elettronici chiave e alta “elettrofilia”, il che suggerisce che dovrebbero interagire agevolmente con partner biologici. Le mappe della superficie elettrica di ciascuna molecola hanno evidenziato regioni fortemente negative attorno agli atomi di ossigeno e zolfo e regioni positive intorno a certi idrogeni—punti naturalmente predisposti per legami a idrogeno quando un farmaco si inserisce nella tasca di una proteina.
Valutare l’incastro nella tasca di CDK2
Il passo successivo è stato il docking virtuale: collocare ogni candidato nella struttura tridimensionale di CDK2 e lasciare che il computer cerchi la migliore vestibilità, come provare chiavi su una serratura. Diverse molecole, in particolare 2b, 2c e 2d, si sono legate a CDK2 più saldamente dello stesso ATP in queste simulazioni. Hanno formato legami a idrogeno cruciali con due residui, Lys33 e Thr14, che si trovano nel cuore della tasca di legame per l’ATP, e si sono disposte anche contro residui amminoacidici idrofobici che rivestono la cavità. Al contrario, lo scaffold di partenza senza sostituzioni specifiche si legava debolmente, sottolineando come cambiamenti sottili nella decorazione chimica dell’anello possano modificare drasticamente le prestazioni.
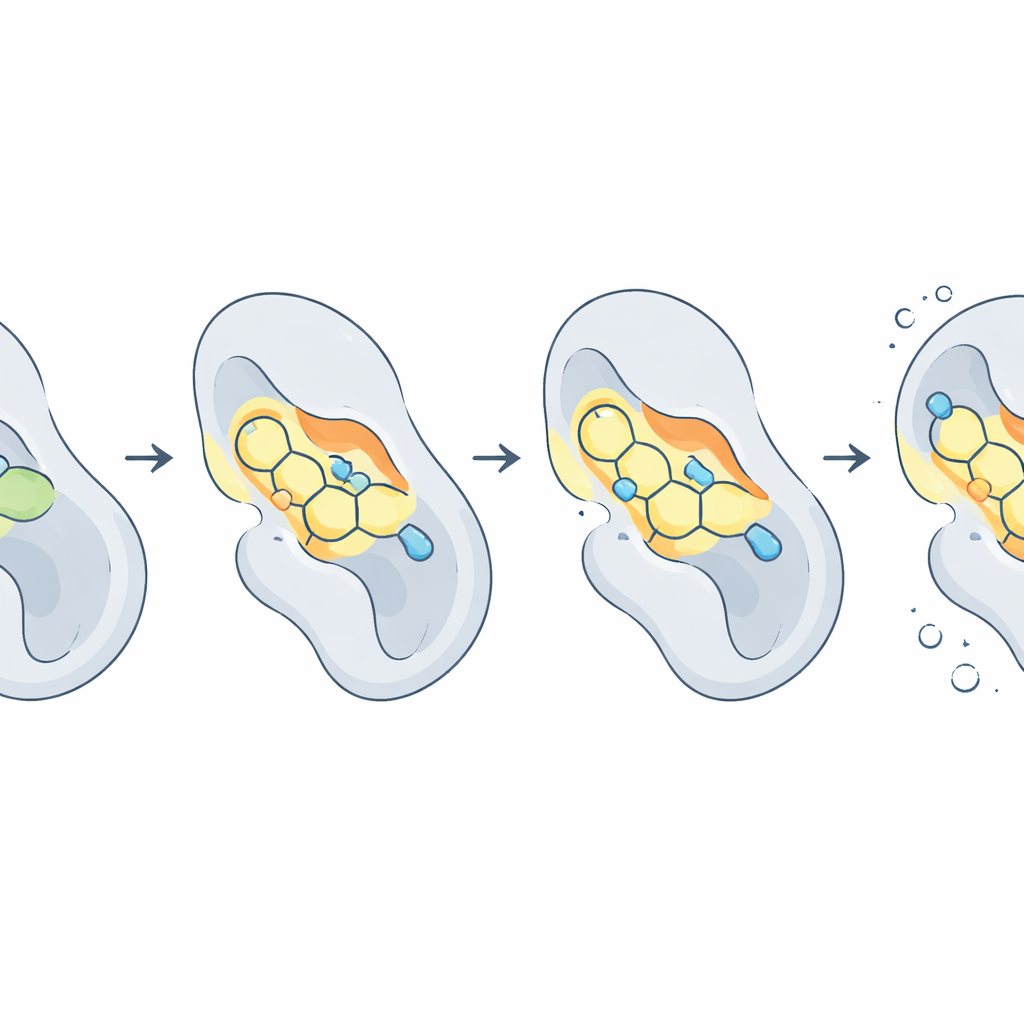
Osservare il complesso proteina–farmaco in movimento
Poiché proteine e farmaci non sono rigidi, gli autori hanno eseguito simulazioni di dinamica molecolare, effettivamente filmati al rallentatore su scala atomica, per decine di nanosecondi. Queste hanno mostrato che la forma complessiva di CDK2 rimaneva stabile mentre alcune coppie proteina–farmaco oscillavano o addirittura si allontanavano. Una molecola, 2f, si è dimostrata eccezionalmente stabile, spostandosi appena nella tasca e mantenendo in media quasi tre legami a idrogeno con la proteina. Altre, come 2c e 2d, hanno bilanciato un legame forte con una flessibilità moderata. Per tradurre questi movimenti in una misura complessiva di “adesività”, il team ha usato una tecnica chiamata MM‑PBSA per stimare le energie libere di legame. In questo contesto, il composto 2d è emerso come il miglior performer a tutto tondo, con un equilibrio particolarmente favorevole tra forze attrattive all’interno della tasca e il costo energetico di distacco dal solvente circostante.
Cosa potrebbe significare per i pazienti
Considerate nel loro insieme, le simulazioni mettono in luce quattro composti di 2‑tioidantoina—2b, 2c, 2d e 2f—come punti di partenza particolarmente promettenti per lo sviluppo di farmaci antitumorali. Combinano proprietà elettroniche favorevoli, un’aderenza forte e ben orientata a CDK2 e, nel caso di 2d, un’energia di legame complessivamente particolarmente vantaggiosa. Pur essendo risultati puramente computazionali che devono essere confermati sperimentalmente, forniscono una mappa dettagliata per i chimici: dove posizionare gruppi alogeno, acido o estere sull’anello centrale per rafforzare legame e selettività. Se i successivi test di laboratorio confermeranno queste previsioni, il lavoro potrebbe accelerare la creazione di terapie mirate su CDK2 più precise, in grado di rallentare la crescita tumorale risparmiando le cellule sane.
Citazione: Khaled, N.A., Ahmed, S.A., Ibrahim, M.A. et al. Integrated DFT, molecular docking, and molecular dynamics investigation of some novel 2-thiohydantoin analogues as potent CDK2 inhibitors for anticancer therapy. Sci Rep 16, 10985 (2026). https://doi.org/10.1038/s41598-026-42330-4
Parole chiave: Inibitori CDK2, Terapia oncologica mirata, Docking molecolare, 2-tioidantoina, Progettazione computazionale di farmaci