Clear Sky Science · fr
La lactylation de l'ALDOB en K87 provoque la fission mitochondriale et le reprogrammation métabolique dans l'hypertension pulmonaire
Pourquoi cela compte pour la santé pulmonaire et cardiaque
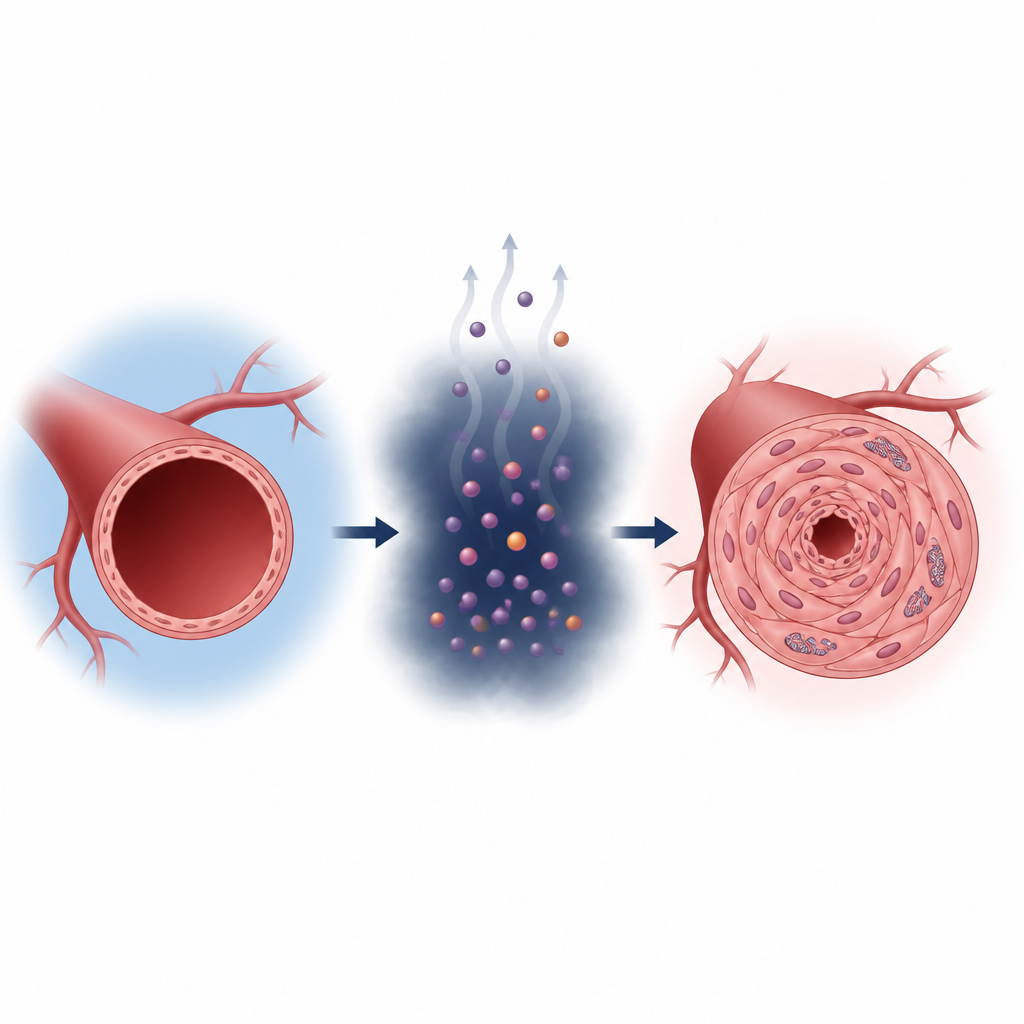
L'hypertension pulmonaire est une maladie grave où les vaisseaux sanguins des poumons se rétrécissent et se rigidifient, sollicitant la partie droite du cœur. De nombreux traitements actuels détendent principalement ces vaisseaux mais n'empêchent guère les lésions sous-jacentes. Cette étude dévoile comment un métabolite courant, le lactate, peut reprogrammer les cellules des vaisseaux pulmonaires et les pousser vers un état pathologique, ouvrant la voie à de nouvelles approches ciblant la cause plutôt que seulement les symptômes.
Du métabolisme du glucose courant aux lésions vasculaires
Les cellules produisent normalement de l'énergie en oxydant les nutriments dans leurs mitochondries, les petites « centrales » cellulaires. Dans l'hypertension pulmonaire, les cellules musculaires lisses qui tapissent les artères pulmonaires basculent vers un mode de production d'énergie plus rapide mais moins efficient, semblable à celui observé dans les cellules cancéreuses. Elles s'appuient davantage sur la glycolyse et génèrent un excès de lactate. Les chercheurs montrent que chez les patients et les rats atteints d'hypertension pulmonaire, les niveaux de lactate et les modifications protéiques liées au lactate dans ces cellules vasculaires sont augmentés, corrélant avec un épaississement paroi artérielle et une insuffisance du ventricule droit.
Comment le lactate actionne un interrupteur moléculaire
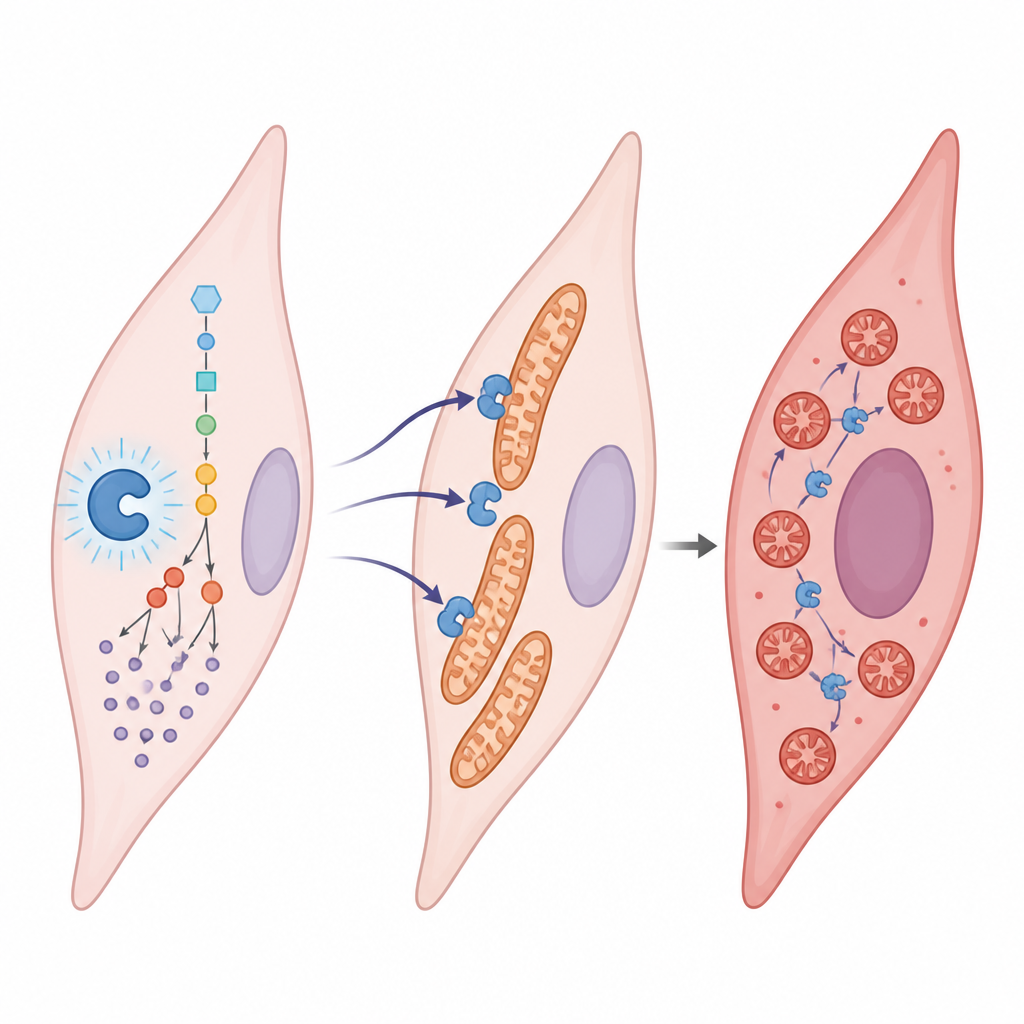
Le lactate est souvent considéré comme un déchet métabolique, mais ici il joue le rôle d'un signal. L'équipe a découvert que le lactate se fixe sur un site spécifique d'une enzyme appelée aldolase B, impliquée dans la dégradation du sucre. Cette marque chimique, portée par un seul acide aminé (K87), augmente l'activité de l'enzyme. L'enzyme accélère alors encore la production de lactate, qui ajoute davantage de marques, créant une boucle auto‑entraînante. Lorsque cette boucle est active en conditions de faible oxygène, les cellules musculaires lisses des artères pulmonaires prolifèrent plus vite, migrent davantage et se transdifférencient en un type plus fibreux et pro‑cicatriciel, favorisant le rétrécissement vasculaire.
Fragmentation des mitochondries au sein des cellules vasculaires
L'étude montre que cette lactylation de l'aldolase B fait plus qu'accélérer la glycolyse : elle remodela aussi les mitochondries. Dans les cellules saines, les mitochondries forment de longs réseaux connectés. Dans les cellules malades, elles se fragmentent en nombreux petits éléments. L'aldolase B lactylée attire une autre protéine, DRP1, vers les mitochondries en modifiant la manière dont DRP1 est modifiée et localisée dans la cellule. Une fois que DRP1 se regroupe à la surface mitochondriale, ces centrales se scindent. Les mitochondries fragmentées sont moins efficaces et soutiennent le comportement agressif et prolifératif observé dans l'hypertension pulmonaire.
Le frein cellulaire et sa défaillance
Les cellules ne sont pas entièrement à la merci de cette boucle entraînée par le lactate. Elles possèdent une enzyme « effaceur », SIRT1, capable d'enlever les marques de lactylation sur des protéines comme l'aldolase B. Les auteurs ont trouvé que les niveaux de SIRT1 sont réduits dans l'hypertension pulmonaire, affaiblissant cette fonction d'effacement. Lorsque SIRT1 était augmenté dans les cellules vasculaires, il retirait la marque de lactylation, calmait le métabolisme glucidique, restaurait des réseaux mitochondriaux plus sains et réduisait la propension des cellules à proliférer et migrer. Chez l'animal, diminuer la lactylation de l'aldolase B ou imiter sa forme non marquée atténuait le remodelage des vaisseaux pulmonaires et la surcharge cardiaque, tandis qu'imiter un état constamment marqué aggravait les traits de la maladie.
Ce que cela signifie pour les traitements futurs
Au total, ce travail cartographie une chaîne causale reliant l'hypoxie, l'excès de lactate, les protéines modifiées, la détérioration mitochondriale et les lésions vasculaires pulmonaires. En termes simples, un axe lactate–aldolase B–DRP1 transforme un dérèglement énergétique en atteinte structurelle des poumons. Pour les patients, cela suggère que des thérapies limitant l'accumulation de lactate, bloquant la marque nocive sur l'aldolase B ou renforçant l'activité effaceuse de SIRT1 pourraient ralentir, voire inverser, les modifications vasculaires responsables de la gravité de l'hypertension pulmonaire, offrant une approche nouvelle au‑delà des simples vasodilatateurs standard.
Citation: Yi, L., He, W., He, C. et al. ALDOB K87 lactylation drives mitochondrial fission and metabolic reprogramming in pulmonary hypertension. Commun Biol 9, 682 (2026). https://doi.org/10.1038/s42003-026-09934-y
Mots-clés: hypertension pulmonaire, métabolisme du lactate, fission mitochondriale, remodelage vasculaire, SIRT1