Clear Sky Science · en
ALDOB K87 lactylation drives mitochondrial fission and metabolic reprogramming in pulmonary hypertension
Why this matters for lung and heart health
Pulmonary hypertension is a serious condition where the blood vessels in the lungs narrow and stiffen, straining the right side of the heart. Many current drugs mainly relax these vessels but do little to stop the underlying damage. This study uncovers how a common metabolic byproduct, lactate, can rewire lung vessel cells and push them into a disease-driving state, pointing to new ways to treat the root cause instead of just the symptoms.
From everyday sugar burning to vessel damage
Cells in our body normally make energy by carefully burning nutrients in their mitochondria, the tiny “power plants” inside each cell. In pulmonary hypertension, smooth muscle cells that line lung arteries shift toward a faster but less efficient style of energy making, similar to cancer cells. They rely more on sugar breakdown and churn out extra lactate. The researchers show that people and rats with pulmonary hypertension have higher lactate levels and more lactate-linked protein changes inside these vessel cells, which go hand in hand with thicker artery walls and a struggling right heart.
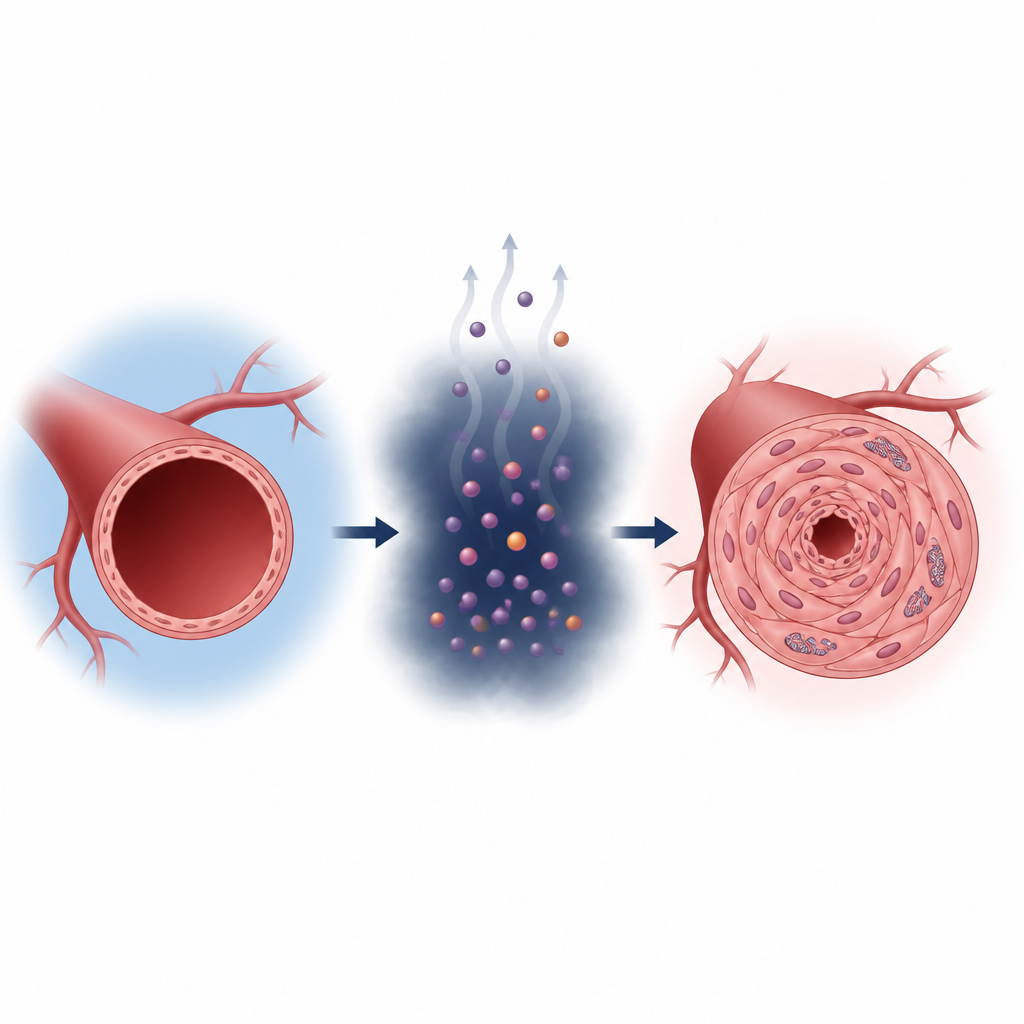
How lactate flips a molecular switch
Lactate is often viewed as metabolic waste, but here it acts more like a signal. The team found that lactate attaches to a specific spot on an enzyme called aldolase B, which helps break down sugar. This chemical tag, placed on a single amino acid (called K87), boosts the enzyme’s activity. The faster enzyme makes even more lactate, which in turn adds more tags, creating a self-reinforcing loop. When this loop is active under low-oxygen conditions, smooth muscle cells in lung arteries grow faster, migrate more, and change into a more fibrous, scar-forming type, all of which promote vessel narrowing.
Breaking mitochondria apart inside vessel cells
The study shows that this lactate tag on aldolase B does more than speed up sugar use; it also reshapes mitochondria. In healthy cells, mitochondria form long, connected networks. In diseased cells, they break into many small fragments. The tagged aldolase B draws another protein, DRP1, to the mitochondria by changing how DRP1 is modified and where it sits in the cell. Once DRP1 clusters on the mitochondrial surface, these power plants split apart. Fragmented mitochondria are less efficient and support the aggressive, growth-prone behavior seen in pulmonary hypertension.
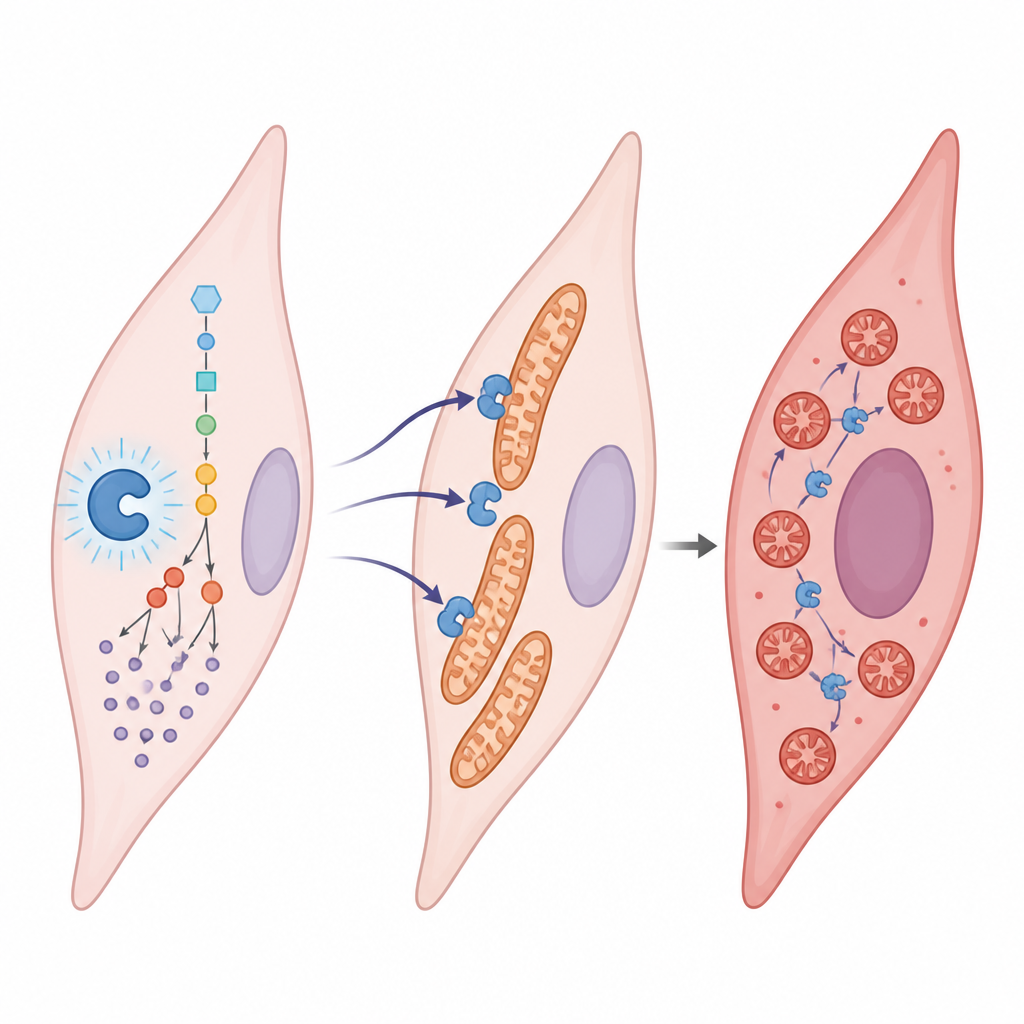
The cell’s own brake and how it fails
Cells are not entirely at the mercy of this lactate-driven loop. They possess a built-in “eraser” enzyme, SIRT1, which can remove lactate tags from proteins like aldolase B. The authors found that SIRT1 levels are reduced in pulmonary hypertension, weakening this eraser function. When SIRT1 was boosted in vessel cells, it peeled off the lactate tag, calmed sugar metabolism, restored healthier mitochondrial networks, and reduced the cells’ tendency to grow and migrate. In animals, dialing down aldolase B lactylation or mimicking the untagged form eased lung vessel remodeling and heart strain, while mimicking the constantly tagged state made disease features worse.
What this means for future treatments
Altogether, the work maps out a chain linking low oxygen, excess lactate, altered proteins, broken mitochondria, and damaged lung vessels. In simple terms, a lactate–aldolase B–DRP1 axis turns energy mismanagement into structural injury in the lungs. For patients, this suggests that therapies which curb lactate buildup, block the harmful tag on aldolase B, or enhance SIRT1’s eraser activity could help slow or even reverse the vessel changes that make pulmonary hypertension so deadly, offering a fresh angle beyond standard vessel-relaxing drugs.
Citation: Yi, L., He, W., He, C. et al. ALDOB K87 lactylation drives mitochondrial fission and metabolic reprogramming in pulmonary hypertension. Commun Biol 9, 682 (2026). https://doi.org/10.1038/s42003-026-09934-y
Keywords: pulmonary hypertension, lactate metabolism, mitochondrial fission, vascular remodeling, SIRT1