Clear Sky Science · es
La lactilación de ALDOB K87 impulsa la fisión mitocondrial y la reprogramación metabólica en la hipertensión pulmonar
Por qué importa para la salud pulmonar y cardiaca
La hipertensión pulmonar es una enfermedad grave en la que los vasos sanguíneos de los pulmones se estrechan y endurecen, forzando al lado derecho del corazón. Muchos fármacos actuales se limitan a relajar estos vasos pero no logran frenar el daño subyacente. Este estudio desvela cómo un subproducto metabólico común, el lactato, puede reprogramar las células de los vasos pulmonares y empujarlas hacia un estado que impulsa la enfermedad, lo que apunta a nuevas formas de tratar la causa raíz en lugar de solo los síntomas.
Del consumo cotidiano de glucosa al daño vascular
Las células de nuestro cuerpo normalmente obtienen energía quemando nutrientes en sus mitocondrias, las pequeñas “centrales eléctricas” dentro de cada célula. En la hipertensión pulmonar, las células musculares lisas que recubren las arterias pulmonares cambian hacia un modo de producción de energía más rápido pero menos eficiente, similar al de las células cancerosas. Dependen más de la degradación de la glucosa y producen lactato en exceso. Los investigadores muestran que personas y ratas con hipertensión pulmonar tienen niveles más altos de lactato y más modificaciones proteicas ligadas al lactato dentro de estas células vasculares, lo que se correlaciona con paredes arteriales más gruesas y un corazón derecho en dificultad.
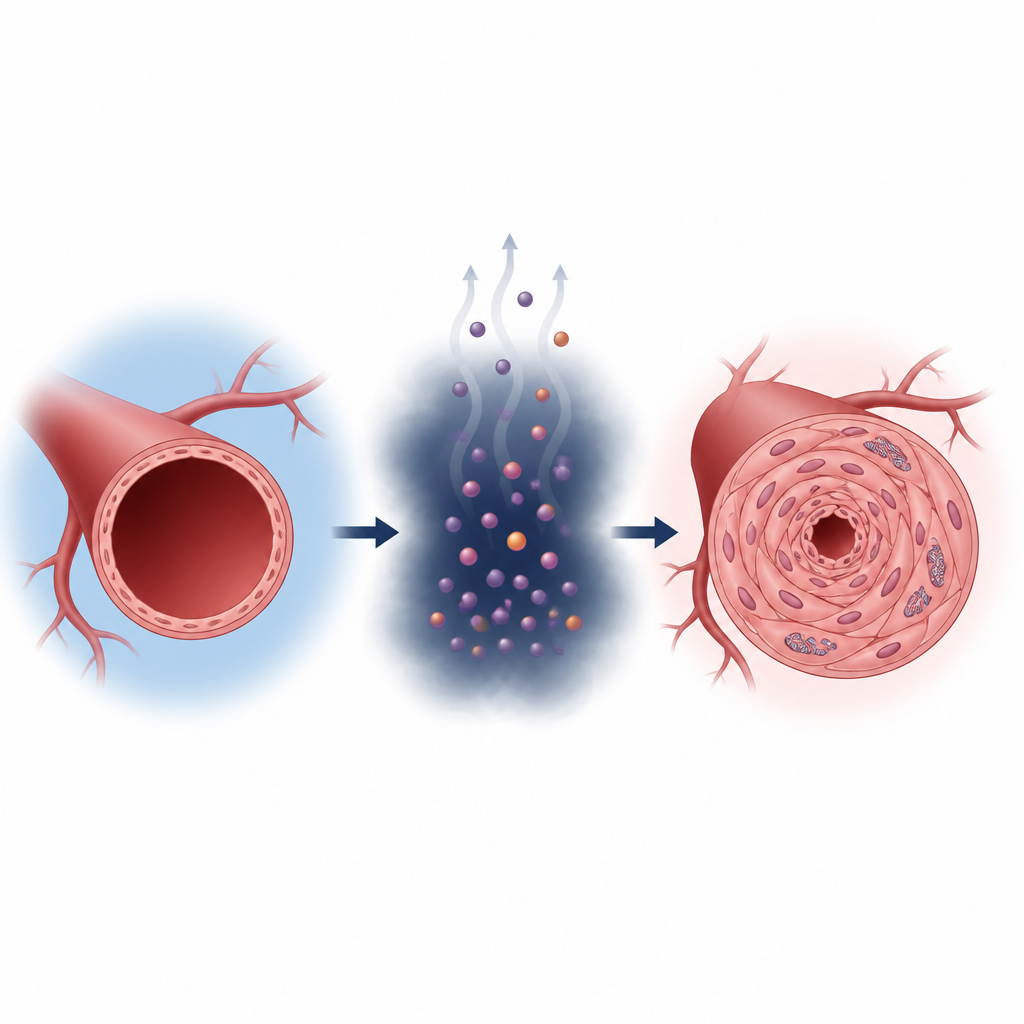
Cómo el lactato cambia un interruptor molecular
El lactato suele considerarse un residuo metabólico, pero aquí actúa más bien como una señal. El equipo encontró que el lactato se une a un punto específico de una enzima llamada aldolasa B, que participa en la degradación de la glucosa. Esta señal química, colocada en un único aminoácido (denominado K87), aumenta la actividad de la enzima. Cuanto más activa es la enzima, más lactato se produce, lo que a su vez añade más marcas, creando un ciclo autorreforzado. Cuando este circuito está activo en condiciones de bajo oxígeno, las células musculares lisas de las arterias pulmonares proliferan más, migran más y se transforman en un tipo más fibrótico y formador de cicatriz, todo lo cual promueve el estrechamiento vascular.
Fragmentando las mitocondrias dentro de las células vasculares
El estudio muestra que esta lactilación de la aldolasa B hace más que acelerar el uso de glucosa; también remodela las mitocondrias. En células sanas, las mitocondrias forman redes largas y conectadas. En las células enfermas, se fragmentan en numerosos pequeños fragmentos. La aldolasa B modificada atrae a otra proteína, DRP1, a las mitocondrias al cambiar cómo se modifica DRP1 y dónde se localiza en la célula. Una vez que DRP1 se agrupa en la superficie mitocondrial, estas centrales se dividen. Las mitocondrias fragmentadas son menos eficientes y sostienen el comportamiento agresivo y proliferativo observado en la hipertensión pulmonar.
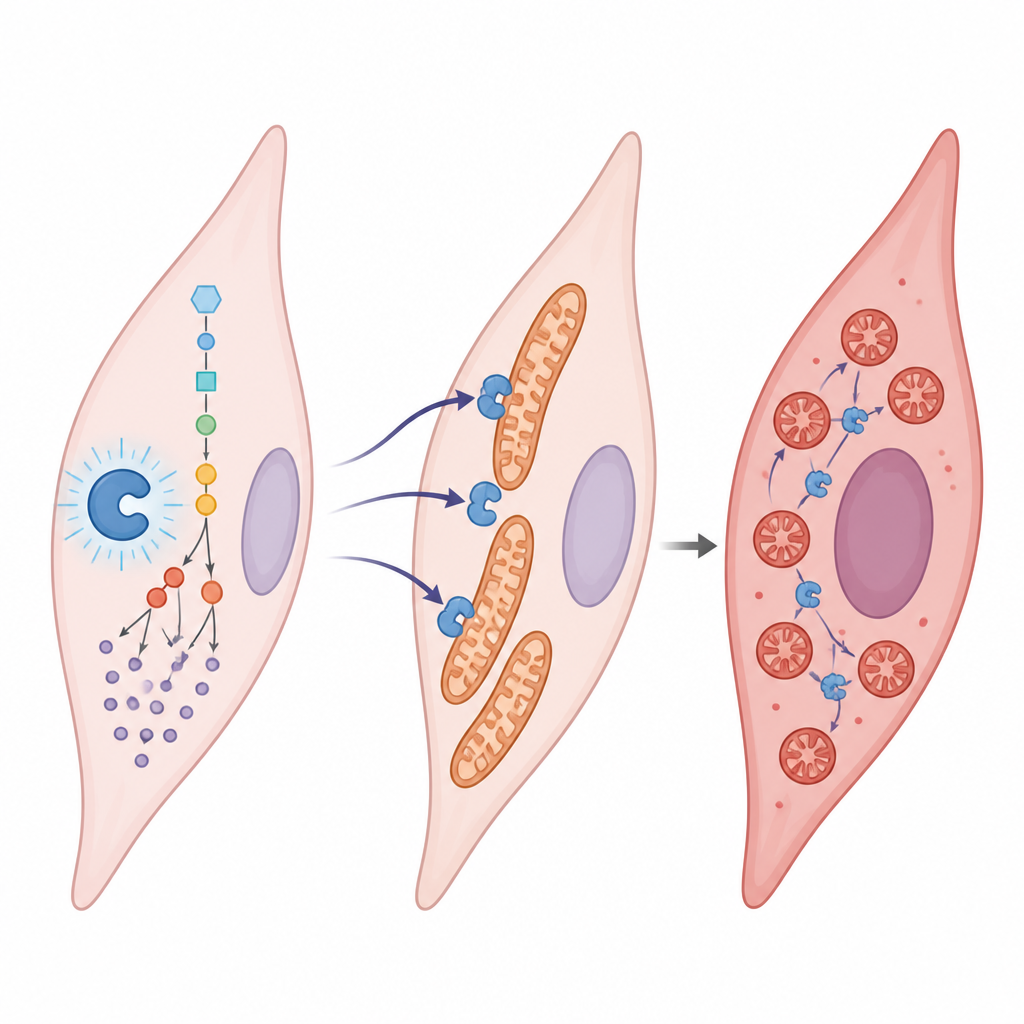
El freno intrínseco de la célula y cómo falla
Las células no están completamente a merced de este circuito impulsado por lactato. Poseen una enzima “borradora” intrínseca, SIRT1, que puede eliminar las marcas de lactato de proteínas como la aldolasa B. Los autores encontraron que los niveles de SIRT1 están reducidos en la hipertensión pulmonar, debilitando esta función de borrado. Cuando SIRT1 se aumentó en las células vasculares, eliminó la marca de lactato, serenó el metabolismo de la glucosa, restauró redes mitocondriales más sanas y redujo la tendencia de las células a proliferar y migrar. En animales, disminuir la lactilación de la aldolasa B o imitar la forma no marcada alivió el remodelado de los vasos pulmonares y la sobrecarga cardiaca, mientras que imitar el estado constantemente marcado empeoró los rasgos de la enfermedad.
Qué implica esto para tratamientos futuros
En conjunto, el trabajo traza una cadena que conecta el bajo oxígeno, el exceso de lactato, proteínas alteradas, mitocondrias fragmentadas y vasos pulmonares dañados. En términos sencillos, un eje lactato–aldolasa B–DRP1 convierte la mala gestión energética en lesión estructural en los pulmones. Para los pacientes, esto sugiere que terapias que reduzcan la acumulación de lactato, bloqueen la marca dañina sobre la aldolasa B o potencien la actividad borradora de SIRT1 podrían ayudar a frenar o incluso revertir los cambios vasculares que hacen que la hipertensión pulmonar sea tan letal, ofreciendo un enfoque nuevo más allá de los fármacos que únicamente relajan los vasos.
Cita: Yi, L., He, W., He, C. et al. ALDOB K87 lactylation drives mitochondrial fission and metabolic reprogramming in pulmonary hypertension. Commun Biol 9, 682 (2026). https://doi.org/10.1038/s42003-026-09934-y
Palabras clave: hipertensión pulmonar, metabolismo del lactato, fisión mitocondrial, remodelado vascular, SIRT1