Clear Sky Science · fr
Z-Calling : un outil pour la lecture des bases A/Z (2,6-diaminopurine) et la détection de l’ADN dZ à partir de lectures PacBio HiFi
Pourquoi un type d’ADN différent compte
On décrit souvent l’ADN comme le plan universel du vivant, constitué de quatre lettres familières : A, T, C et G. Mais certains virus enfreignent discrètement cette règle en remplaçant la lettre A par un analogue chimique appelé Z, qui rend leur ADN plus résistant et plus difficile à cibler. Cette découverte révèle un niveau caché de diversité génétique aux implications importantes pour les biotechnologies, la médecine et même le stockage de données. Le défi, facile à formuler mais difficile à résoudre, est : comment repérer et cartographier de façon fiable cet ADN inhabituel Z, surtout lorsqu’il est mêlé à de l’ADN ordinaire ? Cette étude présente Z-Calling, un outil computationnel qui rend cela enfin possible en utilisant la technologie de séquençage long déjà disponible.
Une lettre étrange dans l’alphabet génétique
Dans la plupart des organismes, la base adénine (A) s’apparie avec la thymine (T) par deux liaisons hydrogène, contribuant à la stabilité de la double hélice d’ADN. Certains bactériophages — virus infectant les bactéries — ont évolué pour remplacer l’A par la 2,6-diaminopurine, surnommée Z. Z forme trois liaisons hydrogène avec T, rendant l’hélice plus stable et modifiant son comportement physique. Cette chimie inhabituelle peut conférer aux virus contenant Z un avantage face aux défenses de l’hôte et ouvre des possibilités pour des molécules conçues offrant de meilleures performances. Des études ont déjà montré que Z peut affiner des tests génétiques, moduler les réactions CRISPR et réduire les réponses immunitaires aux médicaments à base d’ARN expérimentaux. Pourtant, sans méthode permettant de localiser précisément chaque Z le long d’un brin d’ADN, il a été difficile de comprendre pleinement ou d’exploiter ces bénéfices.
Les outils actuels sont insuffisants
Les technologies de séquençage standard partent du principe que tous les signaux de type A correspondent à de l’adénine, et tendent donc à interpréter Z comme une A ordinaire. Des méthodes chimiques, comme la chromatographie liquide haute performance, peuvent quantifier la quantité de Z dans un échantillon global, mais elles ne renseignent pas sur l’emplacement de chaque Z dans un génome, notamment dans des échantillons environnementaux complexes mêlant de nombreuses espèces. Certains instruments de troisième génération, comme les appareils à nanopores, sont théoriquement sensibles aux différences entre bases, mais en pratique leurs signaux peuvent être bruyants et difficiles à interpréter face à une chimie inhabituelle. Jusqu’à présent, il n’existait pas de méthode pratique et fiable pour balayer des mélanges d’ADN complexes et distinguer clairement l’ADN normal de l’ADN contenant Z, ni pour différencier A et Z lettre par lettre.
Écouter le rythme de la synthèse de l’ADN
Les auteurs se sont concentrés sur le séquençage PacBio Circular Consensus Sequencing, une technologie qui copie plusieurs fois la même molécule d’ADN et enregistre non seulement quelle base est ajoutée, mais aussi la rapidité de chaque ajout. Deux mesures temporelles — la durée d’impulsion (pulse width, combien de temps la polymérase met pour ajouter une base) et la durée entre impulsions (inter-pulse duration, la pause entre deux ajouts) — forment une sorte de trace rythmique de la synthèse de l’ADN. En comparant de nombreux échantillons d’ADN conçus avec soin, incluant de l’ADN ordinaire, de l’ADN entièrement substitué en Z et des molécules hybrides où coexistent A et Z, l’équipe a montré que le remplacement de A par Z induit des changements temporels subtils mais cohérents. Ces changements dépendent du contexte séquentiel environnant et affectent principalement la durée d’impulsion dans une fenêtre étroite autour du site Z, tout en laissant la précision globale du séquençage presque aussi élevée que pour l’ADN non modifié.
Comment Z-Calling détecte les bases Z cachées
S’appuyant sur ces motifs cinétiques, les chercheurs ont entraîné des modèles d’apprentissage automatique pour reconnaître « l’accent » cinétique de Z. Leur outil, Z-Calling, remplit deux fonctions principales. D’abord, il classe des lectures entières comme provenant d’ADN ordinaire ou d’ADN contenant Z, y compris dans des métagénomes artificiels mélangeant de nombreuses espèces et chimies. Il procède en utilisant un réseau de neurones pour attribuer à chaque position de type A un score de probabilité d’être Z, puis en fournissant la distribution de ces scores à une machine à vecteurs de support qui décide si la lecture entière provient d’un ADN Z. Ensuite, il effectue une discrimination au niveau d’une seule lettre, attribuant à chaque position A ou Z selon le contexte séquentiel local et les signaux cinétiques. Sur des jeux de données issus de bactéries, levures, plantes, animaux et d’un phage naturellement porteur de Z, ces modèles ont atteint une grande précision (valeurs d’aire sous la courbe autour de 0,94–0,98), comparable aux meilleurs outils qui détectent des marques courantes de méthylation de l’ADN.
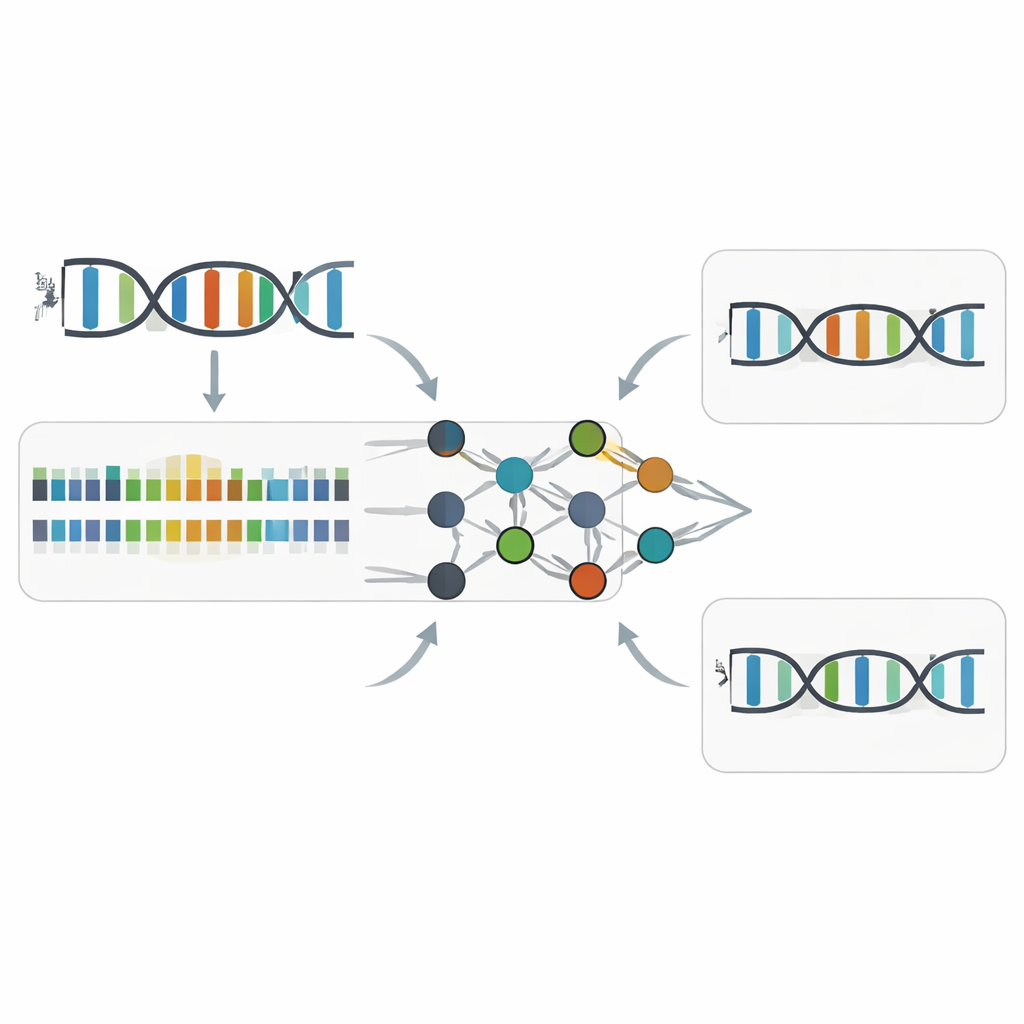
Mettre l’outil à l’épreuve du monde réel
Pour montrer que Z-Calling fonctionne au-delà des constructions propres de laboratoire, les auteurs l’ont appliqué à une souche de levure modifiée qui remplace partiellement l’adénine par Z sur l’ensemble de son génome. L’analyse chimique indiquait qu’environ un quart des positions de type A étaient devenues Z. Z-Calling a estimé indépendamment une fraction similaire et cartographié la distribution de Z sur les chromosomes et les plasmides de la levure, révélant une répartition globalement aléatoire. L’outil a aussi analysé des jeux de données mixtes dans lesquels seule une infime fraction des lectures appartenait à des virus contenant Z ou à des génomes hybrides. Même lorsque les lectures contenant Z ne représentaient qu’environ un pour cent du total, Z-Calling pouvait encore signaler leur présence avec une grande confiance, tout en maintenant un très faible taux de fausses alertes sur de nombreux génomes témoins riches en marques épigénétiques naturelles.
Ce que cela signifie pour l’avenir
En transformant des subtilités temporelles du séquençage de l’ADN en signaux clairs, Z-Calling fournit la première méthode pratique pour cartographier systématiquement où apparaissent les bases Z, base par base et génome par génome. Pour un lecteur général, le message clé est que notre alphabet génétique est plus flexible qu’on ne le pensait, et que nous disposons désormais d’un moyen de lire l’une de ses lettres alternatives les plus intrigantes à haute résolution. Cette capacité aidera les chercheurs à rechercher davantage de virus à base de Z dans la nature, à valider des organismes conçus qui utilisent Z pour plus de stabilité ou de nouvelles fonctions, et à explorer des polymères analogues à l’ADN pour le stockage sécurisé d’informations et des thérapeutiques avancées. En bref, Z-Calling transforme une curiosité chimique obscure en une caractéristique traçable des génomes du monde réel.
Citation: Wu, B., Chen, Y., Zhou, Y. et al. Z-Calling: a tool for A/Z (2,6-diaminopurine) base calling and dZ-DNA detection using PacBio HiFi reads. Commun Biol 9, 594 (2026). https://doi.org/10.1038/s42003-026-09849-8
Mots-clés: ADN Z, bases non canoniques, séquençage PacBio, apprentissage automatique en génomique, génomique des phages