Clear Sky Science · fr
Prédiction à l’échelle du protéome de l’impact fonctionnel des variants faux-sens avec ProteoCast
Pourquoi de minuscules mutations comptent pour la santé et la maladie
Chaque humain porte d’innombrables petites modifications de l’ADN, mais seules certaines de ces altérations perturbent le fonctionnement de nos protéines et contribuent aux maladies. Distinguer les différences bénignes des variantes dangereuses est un défi majeur, d’autant plus que nous pouvons désormais éditer les génomes avec des outils comme CRISPR. Cette étude présente ProteoCast, une méthode computationnelle qui exploite l’histoire même de l’évolution pour prédire quelles substitutions d’une seule lettre dans les protéines sont susceptibles d’avoir un effet, et montre qu’elle peut analyser pratiquement l’ensemble des protéines d’un organisme en une seule fois.

Lire l’empreinte de l’évolution sur les protéines
ProteoCast repose sur une idée simple : si une position particulière d’une protéine a à peine changé pendant des centaines de millions d’années, la modifier aujourd’hui est plus susceptible d’être nuisible. Les auteurs alimentent chaque séquence protéique de la drosophile dans de larges bases de données évolutives pour rassembler des protéines apparentées issues de nombreuses espèces. À partir de ces données, ProteoCast estime à quel point chaque substitution d’acide aminé possible serait perturbatrice à chaque position, créant ainsi un « paysage mutationnel » pour cette protéine. La méthode regroupe ensuite les changements prédits en trois catégories intuitives — neutre, faiblement impactant ou fortement impactant — et étiquette aussi chaque position de la protéine comme tolérante ou sensible aux mutations.
Tester les prédictions à l’échelle d’un animal entier
L’équipe a appliqué ProteoCast à pratiquement l’intégralité du protéome de la drosophile Drosophila melanogaster, couvrant plus de 22 000 formes protéiques et environ 300 millions de mutations faux-sens possibles. Ils ont comparé les prédictions de ProteoCast à près de 400 000 variants génétiques connus, incluant des différences naturelles observées dans des populations de mouches sauvages et consanguines et des mutations étudiées expérimentalement connues pour provoquer une perte de fonction partielle ou une létalité franche. ProteoCast a correctement signalé environ 85 % des mutations létales et 73 % des mutations de perte partielle comme étant modérément ou fortement impactantes, tout en classant la grande majorité des variants de population comme neutres. Autrement dit, le seul motif de conservation évolutive s’est avéré très informatif pour savoir quelles altérations nuisent à la fitness de l’organisme entier.
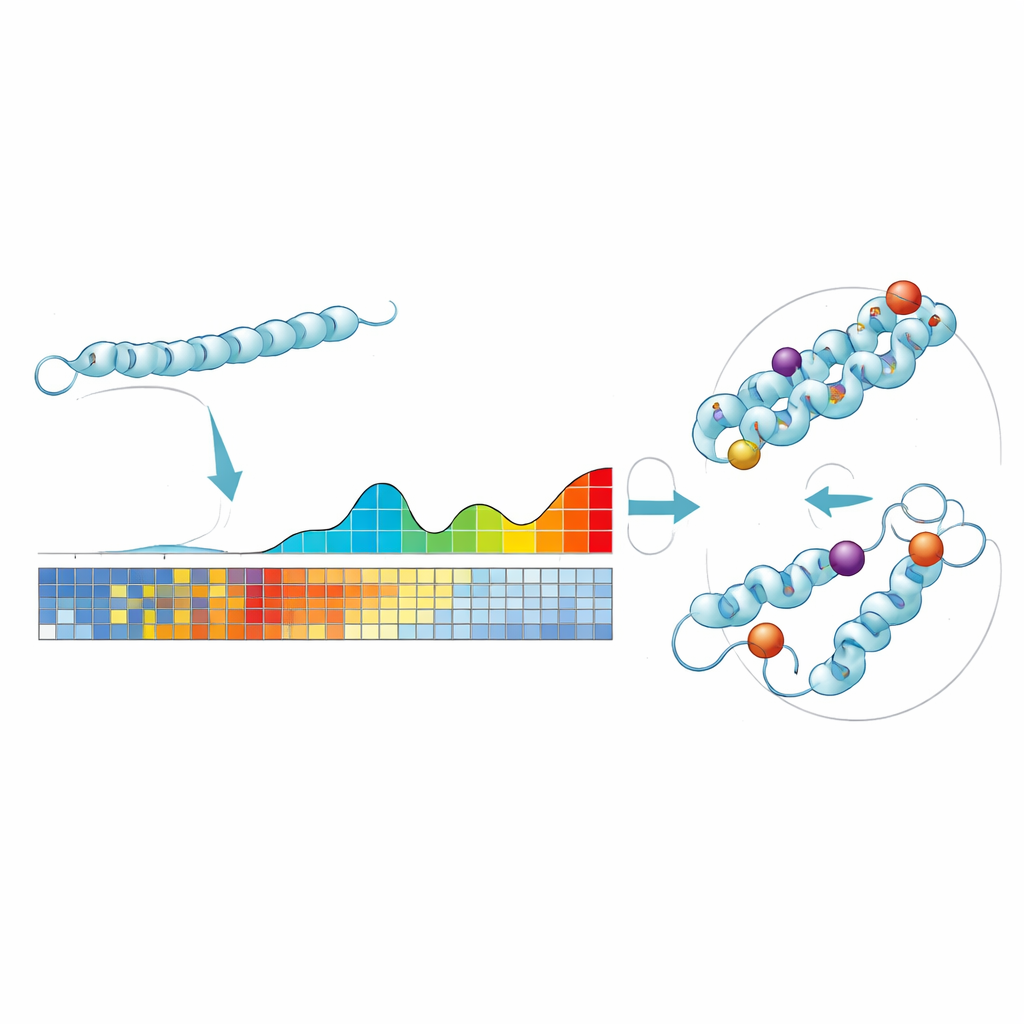
Des scores informatiques à l’édition génomique réelle
Pour vérifier si les résultats de ProteoCast peuvent orienter des expériences, les auteurs s’en sont servis pour choisir des substitutions d’acides aminés spécifiques à éditer par ciblage génomique chez la mouche. Ils se sont concentrés sur une enzyme impliquée dans la production de NAD, un cofacteur métabolique clé. ProteoCast a identifié plusieurs substitutions proches du site actif ou de l’interface de dimérisation de l’enzyme comme fortement impactantes, et d’autres substitutions dans des régions de surface comme neutres, même lorsqu’elles modifiaient radicalement la chimie ou la taille de l’acide aminé. Lorsque ces cinq mutations ont été introduites par CRISPR, les trois prédites comme délétères ont provoqué une létalité développementale récessive, tandis que les deux prédites comme neutres ont donné des mouches saines, en accord avec les prévisions computationnelles.
Trouver des interrupteurs de contrôle cachés dans des régions flexibles
De nombreux sites de régulation importants dans les protéines se trouvent dans des régions « non structurées » qui flottent plutôt que de former des structures 3D stables, ce qui les rend difficiles à étudier. ProteoCast cartographie ses scores de mutation sur des modèles 3D issus d’AlphaFold puis segmente chaque protéine en régions de sensibilité similaire. Les régions où un groupe de positions est anormalement sensible correspondent souvent à des motifs de liaison ou à des points chauds de modifications post‑traductionnelles — des interrupteurs de contrôle subtils qui ajustent l’activité d’une protéine. À l’échelle du protéome de la mouche, les segments à haute sensibilité de ProteoCast chevauchent la plupart des courts motifs linéaires connus et une large fraction des sites de modification, et mettent aussi en évidence des segments non annotés qui participeraient probablement à la régulation ou aux interactions protéine–protéine.
Une portée large au-delà des drosophiles
Bien que le travail se concentre sur la drosophile, le principe derrière ProteoCast est général : l’évolution encode une riche information sur les positions d’une protéine qui peuvent être modifiées sans conséquence et celles qui sont critiques. Les auteurs montrent que le même cadre fonctionne bien sur des variants liés à des maladies humaines et sur des jeux de sites régulateurs validés provenant de la levure et de régions de liaison intrinsèquement désordonnées. Parce qu’il est rapide, scalable et ne nécessite pas de matériel coûteux, ProteoCast peut être appliqué à tout organisme disposant de données de séquences protéiques. Pour les non‑spécialistes, le message clé est que, en laissant l’évolution faire office d’expérimentatrice, nous obtenons une carte puissante, à l’échelle du génome, des minuscules changements génétiques les plus susceptibles d’avoir de l’importance pour la santé, la maladie et les thérapies futures.
Citation: Abakarova, M., Freiberger, M.I., Liehrmann, A. et al. Proteome-wide prediction of the functional impact of missense variants with ProteoCast. Nat Commun 17, 3813 (2026). https://doi.org/10.1038/s41467-026-72140-1
Mots-clés: mutations faux-sens, évolution des protéines, Drosophile, prévision de l’effet des variants, génomique fonctionnelle