Clear Sky Science · en
Proteome-wide prediction of the functional impact of missense variants with ProteoCast
Why tiny mutations matter for health and disease
Every human carries countless small DNA changes, yet only some of these tweaks disrupt how our proteins work and contribute to disease. Sorting harmless differences from dangerous ones is a huge challenge, especially now that we can edit genomes with tools like CRISPR. This study introduces ProteoCast, a computational method that uses the history of evolution itself to predict which single–letter changes in proteins are likely to matter, and shows that it can scan essentially an entire organism’s protein collection at once.

Reading evolution’s fingerprint on proteins
ProteoCast builds on a simple idea: if a particular position in a protein has barely changed over hundreds of millions of years, then altering it today is more likely to be harmful. The authors feed each fruit fly protein sequence into large evolutionary databases to gather related proteins from many species. Using these, ProteoCast estimates how disruptive every possible amino acid substitution would be at every position, creating a “mutational landscape” for that protein. The method then groups predicted changes into three intuitive categories—neutral, mildly impactful, or strongly impactful—and also labels each position in the protein as either tolerant or sensitive to mutation.
Testing predictions across a whole animal
The team applied ProteoCast to virtually the entire proteome of the fruit fly Drosophila melanogaster, covering more than 22,000 protein forms and roughly 300 million possible missense mutations. They compared ProteoCast’s predictions to nearly 400,000 known genetic variants, including natural differences seen in wild and inbred fly populations and experimentally studied mutations known to cause partial loss of function or outright lethality. ProteoCast correctly flagged about 85% of lethal mutations and 73% of partial-loss mutations as mild or impactful, while classifying the vast majority of population variants as neutral. In other words, the pattern of evolutionary conservation alone turned out to be highly informative about which changes hurt the whole organism’s fitness.
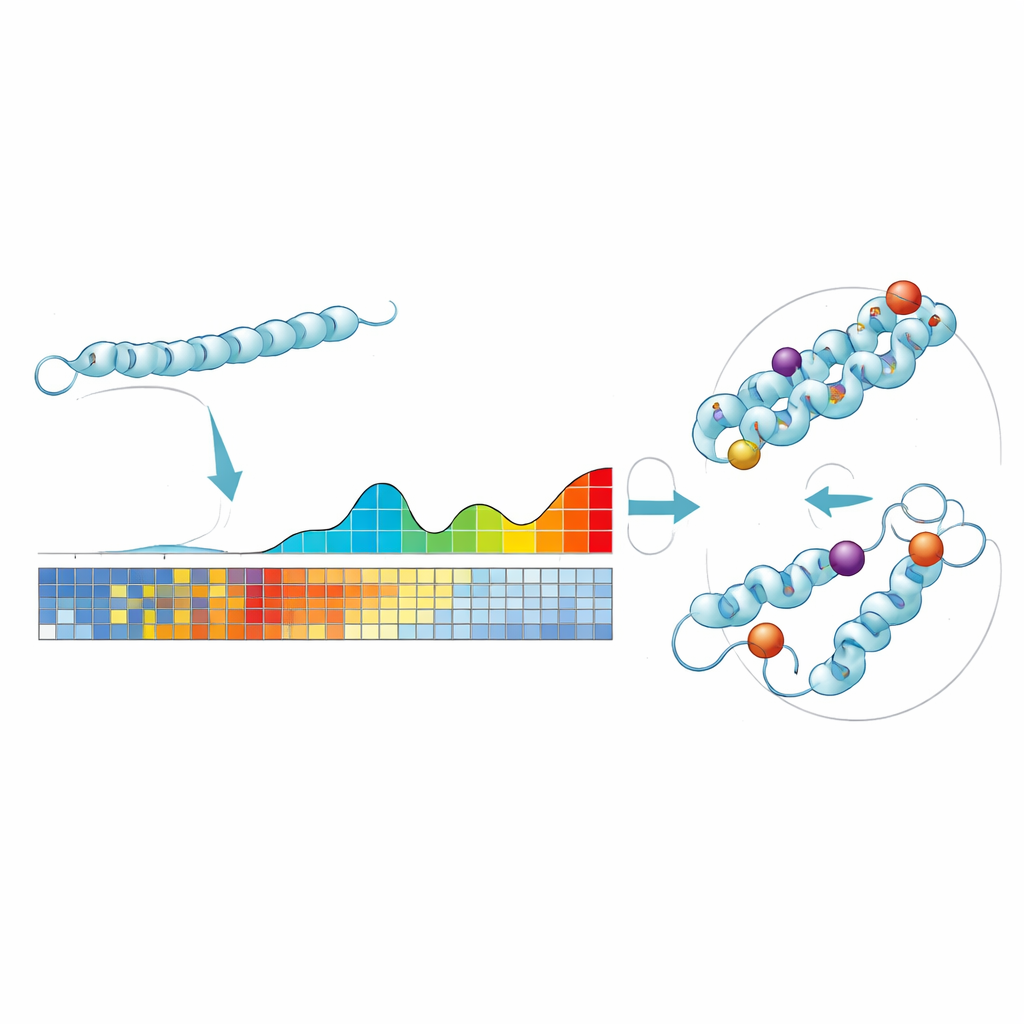
From computer scores to real-life genome editing
To see whether ProteoCast’s output can guide experiments, the authors used it to pick specific single–amino acid changes for targeted genome editing in flies. They focused on an enzyme involved in producing NAD, a key metabolic cofactor. ProteoCast singled out several substitutions near the enzyme’s active or dimer interface as strongly impactful, and other substitutions in surface regions as neutral, even when they drastically changed the chemistry or size of the amino acid. When these five mutations were introduced by CRISPR, the three predicted as damaging caused recessive developmental lethality, whereas the two predicted as neutral yielded healthy flies, matching the computational forecasts.
Finding hidden control switches in floppy regions
Many important regulation sites in proteins lie in “unstructured” regions that flop around rather than forming stable 3D shapes, making them hard to study. ProteoCast maps its mutation scores onto 3D models from AlphaFold and then segments each protein into regions of similar sensitivity. Regions where a cluster of positions is unusually sensitive often correspond to binding motifs or post-translational modification hotspots—subtle control switches that tune a protein’s activity. Across the fly proteome, ProteoCast’s high-sensitivity segments overlapped with most known short linear motifs and a large fraction of modification sites, and they also highlighted previously unannotated segments that likely participate in regulation or protein–protein interactions.
Broad impact beyond fruit flies
Although the work centers on fruit flies, the principle behind ProteoCast is general: evolution encodes rich information about which positions in a protein can be changed without consequence, and which ones are critical. The authors show that the same framework performs well on human disease variants and on curated sets of regulatory sites from yeast and intrinsically disordered binding regions. Because it is fast, scalable, and does not require expensive hardware, ProteoCast can be applied to any organism with protein sequence data. For non-specialists, the key message is that by letting evolution be the experimenter, we gain a powerful, genome-wide map of which tiny genetic changes are most likely to matter for health, disease, and future therapies.
Citation: Abakarova, M., Freiberger, M.I., Liehrmann, A. et al. Proteome-wide prediction of the functional impact of missense variants with ProteoCast. Nat Commun 17, 3813 (2026). https://doi.org/10.1038/s41467-026-72140-1
Keywords: missense mutations, protein evolution, Drosophila, variant effect prediction, functional genomics