Clear Sky Science · fr
Le transporteur d’acides aminés LAT1 coordonne la fonction motrice correcte au stade périnatal
Pourquoi c’est important pour le mouvement et les maladies infantiles
Apprendre à bouger avec fluidité est l’un des principaux défis que doit relever le système nerveux d’un nouveau-né. Cette étude révèle comment une seule protéine qui transporte des acides aminés dans les cellules nerveuses aide les souriceaux nouveau-nés à construire et maintenir le câblage permettant à la moelle épinière de contrôler leurs muscles. Comme des réseaux similaires sont endommagés dans des maladies humaines des neurones moteurs telles que l’amyotrophie spinale, ce travail ouvre une nouvelle manière de penser l’influence de la nutrition précoce et des systèmes de transport cellulaires sur la santé motrice à long terme. 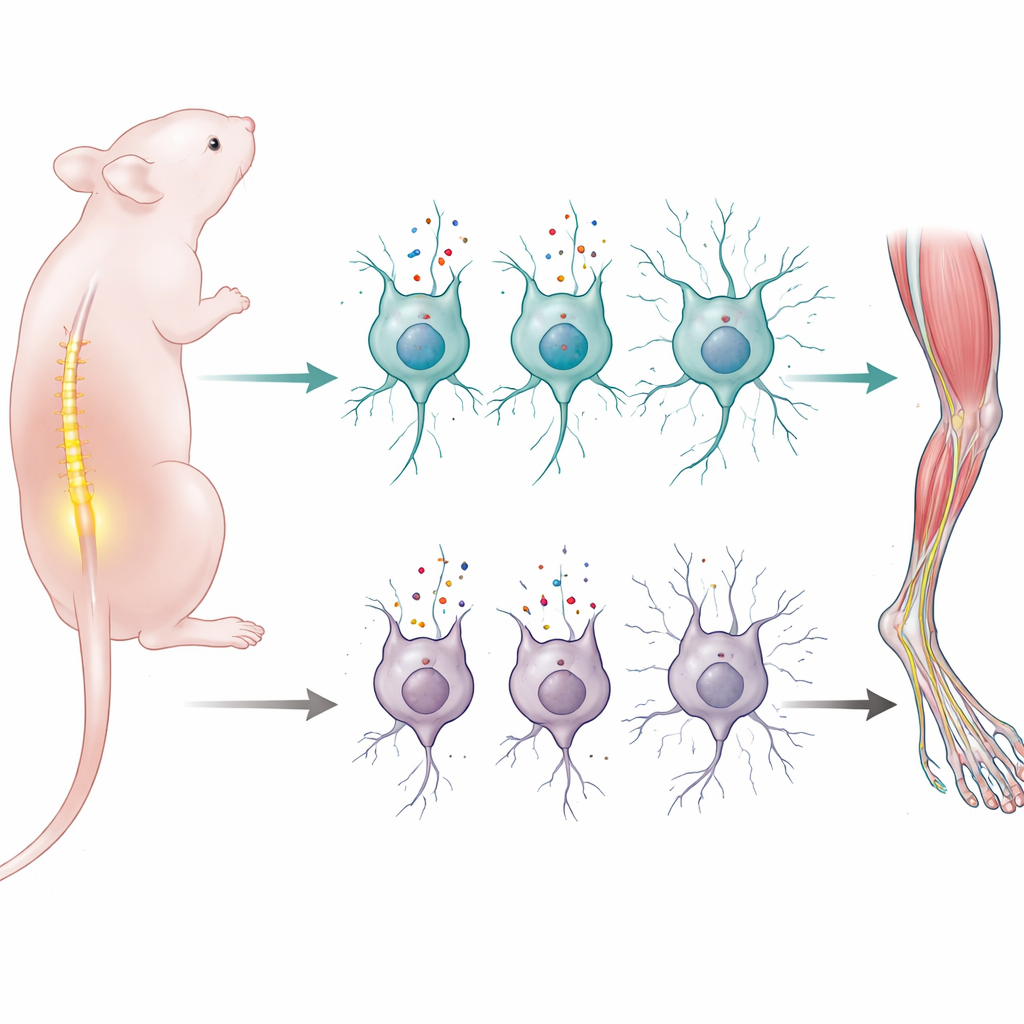
Une porte d’entrée nutritive dans les cellules nerveuses
Les chercheurs se sont concentrés sur une protéine appelée LAT1, qui sert de porte d’entrée pour les grands acides aminés neutres — les éléments de base et le carburant de nombreux processus cellulaires — dans certains neurones. LAT1 n’est pas réparti uniformément dans l’organisme : son expression est fortement activée dans des tissus et à des moments particuliers, notamment autour de la naissance. Des travaux antérieurs avaient montré qu’une suppression complète du gène LAT1 chez la souris entraîne de graves problèmes de développement cérébral et la mort avant ou peu après la naissance, mais on ne savait pas quelles catégories de cellules nerveuses dépendaient absolument de ce transporteur pour fonctionner.
Éteindre LAT1 dans des neurones spécifiques
Pour préciser le rôle de LAT1, l’équipe a généré des souris chez lesquelles le gène LAT1 (Slc7a5) était supprimé uniquement dans les neurones exprimant une protéine neuronale courante, la synapsine 1. Ces souris semblaient normales à la naissance et apparaissaient dans les proportions attendues, ce qui indiquait que la formation précoce du cerveau pouvait encore se dérouler. Mais au cours des deux premières semaines de vie, les souriceaux développèrent d’importants troubles moteurs : faiblesse, maladresse dans des tests d’équilibre et de coordination, et absence de prise de poids normale. Aucune ne survécut au-delà de trois semaines, suggérant une dépendance critique à LAT1 pendant la période périnatale où les circuits moteurs sont en cours de maturation.
Atteinte de la moelle épinière mais cerveau épargné
Lorsque les scientifiques examinèrent le système nerveux au microscope, ils constatèrent que les dommages les plus marqués se situaient dans la partie inférieure de la moelle épinière, où résident les motoneurones qui commandent directement les muscles des membres. À deux semaines d’âge, environ la moitié de ces motoneurones avaient disparu chez les souris mutantes, en particulier les cellules plus volumineuses qui contrôlent typiquement les fibres musculaires puissantes. Des signes d’autodestruction cellulaire et d’une autophagie excessive apparurent encore plus tôt, suggérant une voie de stress menant à la mort cellulaire. Les cellules de soutien environnantes de la moelle — astrocytes, microglie et cellules productrices de myéline — montrèrent aussi de fortes réactions, marque d’une lésion locale. En revanche, le cortex moteur et le cervelet, qui participent également au contrôle du mouvement, semblaient structurellement normaux, sans perte évidente de neurones ni cicatrisation, ce qui souligne que le dommage principal était concentré sur les motoneurones inférieurs. 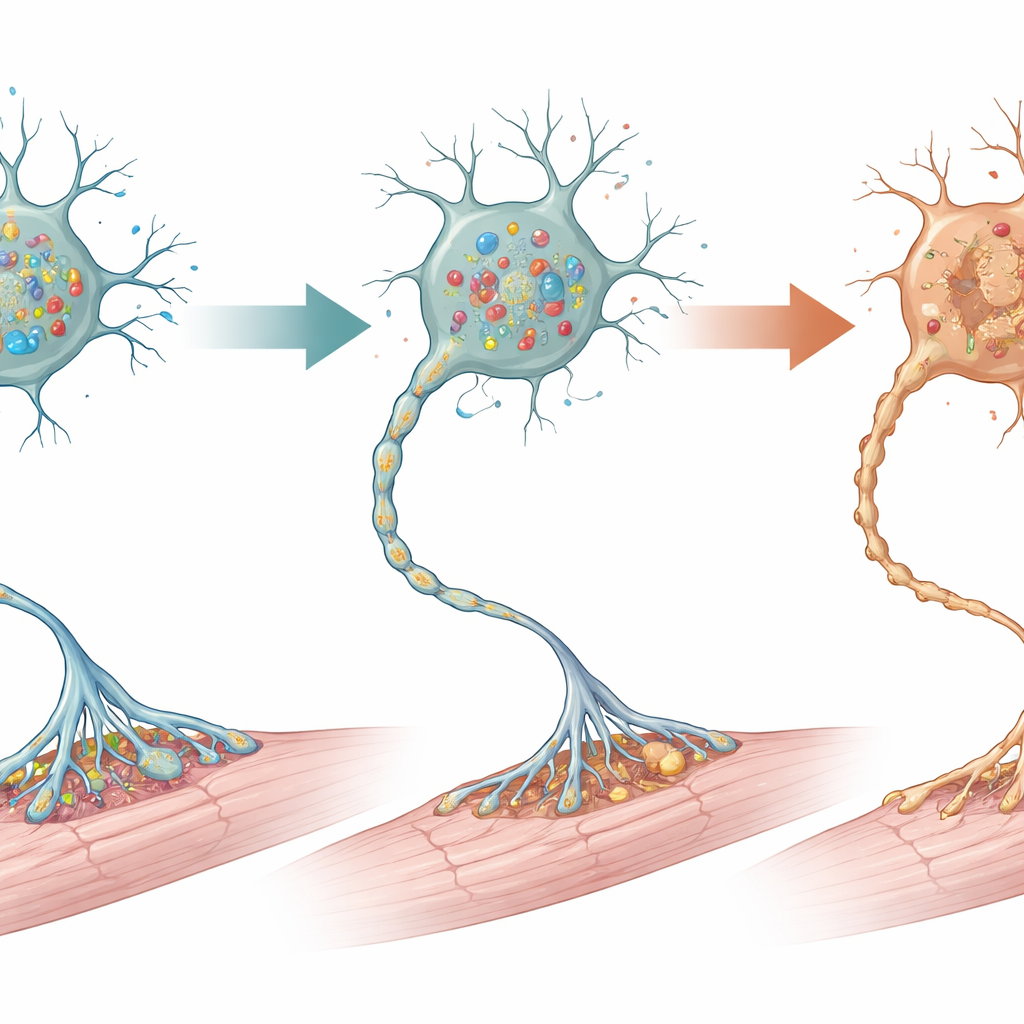
Muscles, jonctions et une sauvegarde partielle
Les conséquences de la perte de motoneurones spinaux se répercutèrent jusqu’aux muscles. Au début, les fibres musculaires paraissaient normales, mais dès la deuxième semaine elles avaient rétréci, signe clair d’atrophie. Les points de connexion nerf–muscle, appelés jonctions neuromusculaires, perdirent leur architecture habituellement complexe et nombreuses devinrent seulement partiellement innervées ou complètement déconnectées de leurs fibres nerveuses. Pour tester si des voies de mort cellulaire conduisaient ce déclin, l’équipe traita les jeunes souris par la calpéptine, un médicament qui atténue une forme de mort cellulaire programmée. Les animaux traités vécurent plus longtemps, conservèrent davantage de motoneurones spinaux et présentaient des jonctions neuromusculaires mieux formées que les mutants non traités, bien que le traitement n’ait pas complètement empêché la maladie.
Liens avec les maladies motrices infantiles
Étant donné que le schéma de perte précoce des motoneurones inférieurs et l’échec des jonctions neuromusculaires rappelait l’amyotrophie spinale, les chercheurs ont également analysé plusieurs grands jeux de données génétiques issus de modèles murins de cette maladie. Dans de multiples études indépendantes, ils observèrent que les gènes impliqués dans le transport et le métabolisme des acides aminés, y compris Slc7a5, étaient régulés à la baisse dans les motoneurones affectés, tandis que les gènes liés à la mort cellulaire et à l’activation gliale étaient régulés à la hausse. Cette convergence suggère qu’un affaiblissement du transport d’acides aminés pourrait être un facteur commun dans les troubles moteurs d’apparition précoce et que préserver ou renforcer la fonction de LAT1 dans les motoneurones spinaux vulnérables pourrait constituer une stratégie prometteuse pour diagnostiquer ou traiter ces affections.
Message clé pour la santé motrice
En termes simples, ce travail montre que certains motoneurones spinaux chez les nouveau-nés dépendent d’une « porte » spécialisée d’apport en acides aminés pour survivre et former des connexions solides avec le muscle. Lorsque cette porte est fermée par la suppression de LAT1, les neurones manquent de nutriments clés, subissent un stress et meurent, entraînant faiblesse, fonte musculaire et mort précoce. En reliant cette voie aux schémas observés dans des modèles d’amyotrophie spinale, l’étude met en lumière le transport d’acides aminés — et la protéine LAT1 en particulier — comme un levier potentiel pour comprendre et, à terme, intervenir dans des troubles moteurs sévères de la petite enfance.
Citation: Sadamori, K., Hiraiwa, M., Horie, T. et al. The amino acid transporter LAT1 coordinates proper motor function at the perinatal stage. Cell Death Dis 17, 345 (2026). https://doi.org/10.1038/s41419-026-08663-8
Mots-clés: neurones moteurs, transport d’acides aminés, LAT1, amyotrophie spinale, jonction neuromusculaire