Clear Sky Science · en
The amino acid transporter LAT1 coordinates proper motor function at the perinatal stage
Why this matters for movement and childhood disease
Learning to move smoothly is one of the biggest jobs a newborn’s nervous system has to master. This study uncovers how a single protein that ferries amino acids into nerve cells helps newborn mice build and maintain the wiring that lets their spinal cord control their muscles. Because similar wiring is damaged in human motor neuron diseases like spinal muscular atrophy, the work points to a new way of thinking about how early-life nutrition and cellular transport systems can influence long-term motor health. 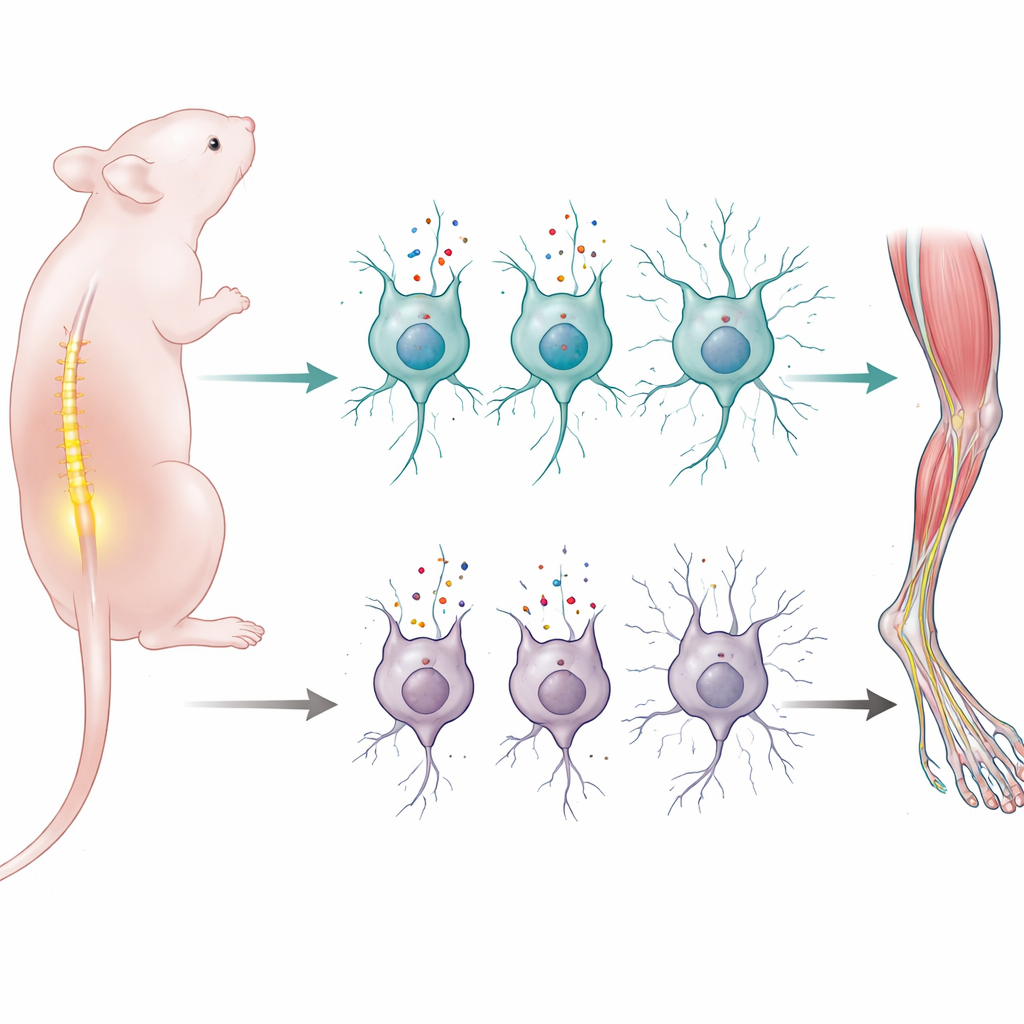
A nutrient gateway in nerve cells
The researchers focused on a protein called LAT1, which acts as a gateway for large neutral amino acids—the building blocks and fuel for many cell processes—into certain nerve cells. LAT1 is not spread evenly throughout the body: it is turned on strongly in particular tissues and at particular times, including around birth. Past work had shown that completely removing the LAT1 gene in mice causes severe brain development problems and death before or shortly after birth, but it was not clear which nerve cell types absolutely depended on this transporter to function.
Switching off LAT1 in specific neurons
To pinpoint LAT1’s role, the team bred mice in which the LAT1 gene (Slc7a5) was removed only in neurons that express a common nerve-cell protein called synapsin 1. These mice appeared normal at birth and were born in expected numbers, which meant that early brain formation could still occur. But within the first two weeks of life, the young mice developed striking motor problems: they were weak, clumsy in tests that measured balance and coordination, and failed to gain normal weight. None survived beyond three weeks, suggesting a critical dependence on LAT1 during the perinatal period when motor circuits are being refined.
Spinal cord damage but a spared brain
When the scientists examined the nervous system under the microscope, they found that the most dramatic damage was in the lower spinal cord, where motor neurons that directly drive limb muscles reside. By two weeks of age, about half of these motor neurons were gone in the mutant mice, especially the larger cells that typically control powerful muscle fibers. Signs of cell self-destruction and excessive cellular “self-cleaning” (autophagy) appeared even earlier, hinting at a stress pathway leading to cell death. Surrounding support cells in the spinal cord—astrocytes, microglia, and myelin-producing cells—also showed strong reactive changes, a hallmark of local injury. In contrast, the motor cortex and cerebellum in the brain, which also participate in movement control, looked structurally normal, with no obvious loss of neurons or scarring, underscoring that the key damage was concentrated in lower motor neurons. 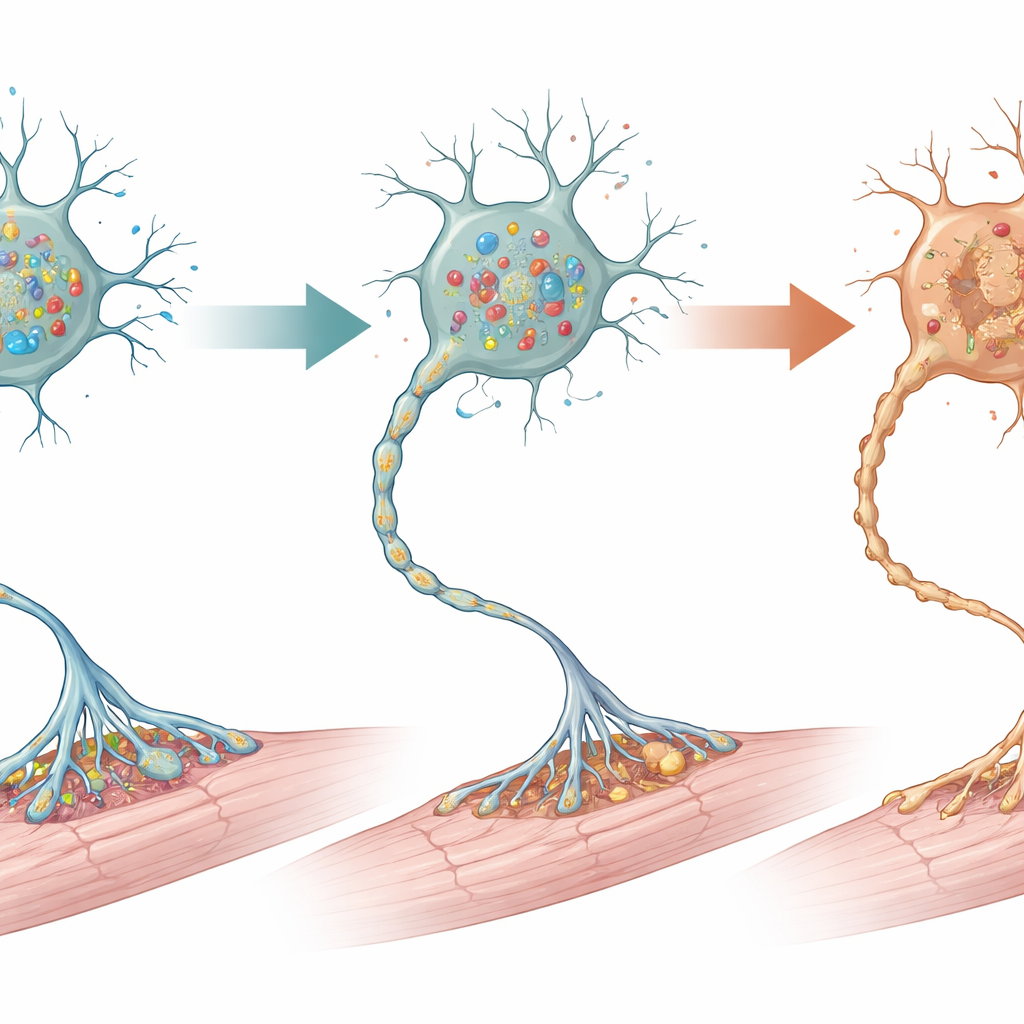
Muscles, junctions, and a partial rescue
The consequences of spinal motor neuron loss extended outward to the muscles. Early on, muscle fibers looked normal, but by the second week they had shrunk, a clear sign of atrophy. The nerve–muscle connection points, called neuromuscular junctions, lost their usual intricate shape and many became only partially supplied or completely disconnected from their nerve fibers. To test whether cell death pathways were driving this decline, the team treated young mice with calpeptin, a drug that dampens a form of programmed cell death. Treated animals lived longer, retained more spinal motor neurons, and had better-formed neuromuscular junctions than untreated mutants, although the treatment did not completely prevent disease.
Links to childhood motor neuron disease
Because the pattern of early lower motor neuron loss and failing neuromuscular junctions resembled spinal muscular atrophy, the researchers also combed through several large genetic datasets from mouse models of that disease. Across multiple independent studies, they found that genes involved in amino acid transport and metabolism, including Slc7a5, were dialed down in affected motor neurons, and genes linked to cell death and glial activation were dialed up. This convergence suggests that weakened amino acid transport may be a common thread in early-onset motor neuron disorders and that preserving or boosting LAT1 function in vulnerable spinal motor neurons could be a promising future strategy to diagnose or treat such conditions.
Take-home message for movement health
In simple terms, this work shows that certain spinal motor neurons in newborn mice depend on a specialized amino acid “supply door” to survive and form strong connections with muscle. When that door is shut by removing LAT1, the neurons run short of key nutrients, become stressed, and die, leading to weak, wasting muscles and early death. By tying this pathway to patterns seen in spinal muscular atrophy models, the study highlights amino acid transport—and the LAT1 protein in particular—as a potential new handle for understanding and eventually intervening in severe early-life movement disorders.
Citation: Sadamori, K., Hiraiwa, M., Horie, T. et al. The amino acid transporter LAT1 coordinates proper motor function at the perinatal stage. Cell Death Dis 17, 345 (2026). https://doi.org/10.1038/s41419-026-08663-8
Keywords: motor neurons, amino acid transport, LAT1, spinal muscular atrophy, neuromuscular junction