Clear Sky Science · de
Antisense-Oligonukleotid-vermittelte Knockdown-Therapie bei zwei Säuglingen mit schwerer KCNT1-epileptischer Enzephalopathie
Warum diese Studie für Familien wichtig ist
Einige der verheerendsten Epilepsien beginnen in den ersten Lebenstagen und sprechen nicht auf Standardmedikamente an, sodass Säuglinge dauerhafte Anfälle und schwere Entwicklungsbeeinträchtigungen erleiden. Diese Studie untersucht eine hochgradig personalisierte Form der Genmedizin – ein Antisense-Oligonukleotid-Medikament –, das entwickelt wurde, um die Aktivität eines einzelnen überaktiven Gehirnkanals bei zwei kleinen Mädchen herunterzuregeln. Sie gewährt einen Einblick, wie Präzisionstherapien ansonsten unbehandelbare Anfälle zügeln könnten, zeigt aber zugleich ernsthafte Risiken auf, die verstanden werden müssen, bevor solche Ansätze breit angewendet werden.
Das Problem unaufhörlicher Anfälle im frühen Leben
Die infantile Epilepsie mit wandernden fokalen Anfällen ist eine seltene, aber extrem schwere Erkrankung. Babys entwickeln Anfälle innerhalb von Stunden oder Tagen nach der Geburt und erleben trotz vielfältiger Antiepileptika oft dutzende Anfälle pro Tag. Bei etwa der Hälfte dieser Kinder liegt die Ursache in einer neuen Mutation im Gen KCNT1, das einen Kanal codiert, der Kaliumionen durch die Membran von Nervenzellen lässt. Die hier untersuchte spezifische Mutation, p.R474H, macht die Kanäle deutlich aktiver als normal und treibt neuronale Netzwerke in einen dauerhaft übererregten Zustand. Diese Kinder zeigen typischerweise kaum Entwicklungsschritte, haben ein hohes Risiko für frühzeitigen Tod und derzeit keine verlässlich wirksamen Behandlungen.
Ein im Labor entwickeltes genzielgerichtetes Medikament
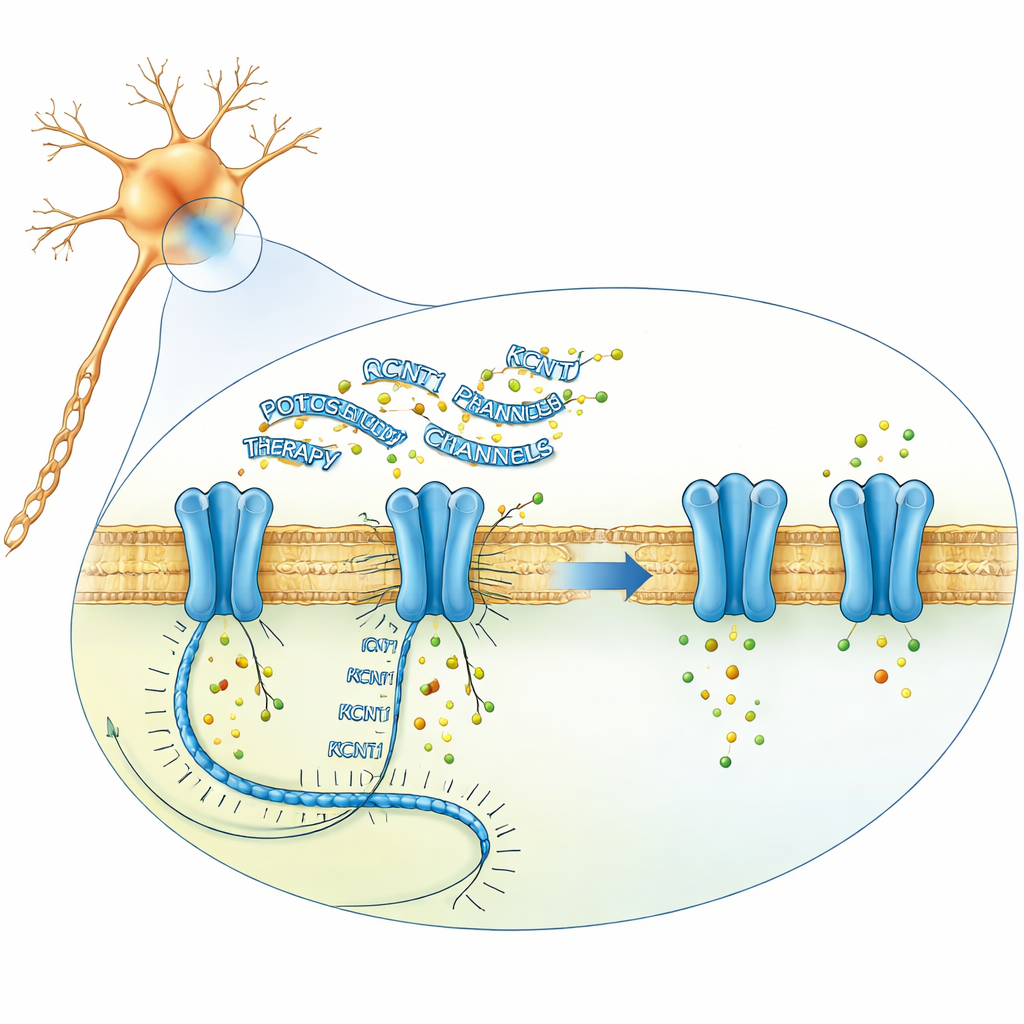
Das Forscherteam wollte die Menge der fehlerhaften KCNT1-Botschaft in Neuronen mithilfe von Antisense-Oligonukleotiden reduzieren: kurzen, chemisch modifizierten Strängen genetischen Materials, die an eine ausgewählte RNA binden und deren Abbau auslösen können. Aus mehr als 250 Kandidatensequenzen selektierten sie schrittweise Designs in kultivierten menschlichen nervenähnlichen Zellen, in aus Patientenstammzellen gezüchteten Neuronen und in Mauszellen. Ihre führende Verbindung, später Valeriasen genannt, senkte KCNT1-RNA- und Proteinspiegel in patientenabgeleiteten Neuronen und in einer Mäuselinie mit der entsprechenden Krankheitsmutation stark, ohne breit andere Gene zu stören. Elektrophysiologische Experimente zeigten, dass das Medikament die ungewöhnlich großen Kaliumströme normalisierte und das Feuermuster der mutierten Neuronen deutlich näher an das gesunder Zellen brachte, was darauf hindeutet, dass eine Abschwächung des Kanals das zugrundeliegende elektrische Ungleichgewicht korrigieren könnte.
Tests in Tiermodellen vor der Anwendung am Menschen
Bevor ein Einsatz bei Kindern erwogen wurde, bewerteten die Forschenden das Medikament in mehreren Nager-Modellen. In Mäusen, die menschliches KCNT1 exprimieren, reduzierten Injektionen in die Gehirnflüssigkeit sowohl Maus- als auch menschliche KCNT1-RNA dosisabhängig. In einer anderen Mausstamm, die aufgrund einer anderen KCNT1-Mutation natürlicherweise schwere Anfälle, neurokognitive Probleme und frühen Tod entwickelt, verlängerte die Behandlung mit dem führenden Wirkstoff die mittlere Lebensdauer von etwa sechs Wochen auf mehrere Monate und verbesserte einfache Verhaltensmaße wie Nestbau. Studien an Ratten mit wiederholten Spinalinjektionen halfen, Dosen zu bestimmen, die nach Verhaltensbeobachtung, neurologischer Untersuchung und Gewebsanalyse allgemein verträglich erschienen und dennoch eine beträchtliche KCNT1-Reduktion in Gehirnregionen erreichten. Diese Ergebnisse unterstützten den vorsichtigen, eng überwachten Einsatz bei Patienten im Rahmen eines besonderen Prüfverfahrens.
Frühe Erfahrungen bei zwei Kindern
Zwei drei Jahre alte Mädchen mit der KCNT1 p.R474H-Mutation, beide mit Anfällen seit dem Neugeborenenalter und minimaler Entwicklungsfortschritt, erhielten steigende Dosen von Valeriasen, verabreicht per Lumbalpunktion in die Flüssigkeit rund um das Rückenmark. Bei beiden gingen die von Familien aufgezeichneten und durch wiederholte Elektroenzephalogramme bestätigten Anfallzahlen nach mehreren Dosen deutlich zurück, und ein Kind zeigte Verbesserungen bei Kopfkontrolle, Schlucken und der Fähigkeit, sich selbst zu beruhigen. Die Medikamentenspiegel in Liquor und Blut verhielten sich wie in den Tierversuchen vorhergesagt. Nach Monaten der Behandlung mit relativ hohen kumulativen Dosen entwickelten jedoch beide Kinder eine schwerwiegende Komplikation: eine progrediente Vergrößerung der mit Flüssigkeit gefüllten Hirnräume mit erhöhtem Druck, eine Form des Hydrozephalus. Die Familie eines Kindes entschied sich für palliative Versorgung; das Kind starb später. Das andere Kind benötigte eine operativ eingesetzte Drainage (Shunt), um überschüssige Flüssigkeit abzuführen.
Abwägung von Hoffnung und Risiko
Angesichts dieser unerwarteten Nebenwirkung unterbrach das Team die Gabe und prüfte Tier- und Labordaten sorgfältig. Hydrozephalus war in Mäusemodellen ohne KCNT1 oder mit Krankheitsmutationen nicht aufgetreten, und genetische Analysen legen nahe, dass Menschen das Verlieren einer KCNT1-Kopie tolerieren können, was gegen eine einfache On-Target-Toxizität spricht. Einige andere Antisense-Präparate, die bei neurologischen Erkrankungen eingesetzt werden, wurden mit seltenen Fällen von erhöhtem Hirndruck oder Ventrikelvergrößerung in Verbindung gebracht, besonders bei höheren Dosen, was andeutet, dass das Problem von Wechselwirkungen bestimmter Verbindungen mit den flüssigkeitsproduzierenden und -ableitenden Strukturen des Gehirns herrühren könnte. Nach einer zweijährigen Pause setzte die zweite Patientin die Behandlung nach einem überarbeiteten, niedrigerdosierten Plan mit zusätzlichen MRT-Untersuchungen und Druckkontrollen fort; unter diesem vorsichtigen Regime erlebte sie erneut etwa eine um zwei Drittel verringerte Anfallshäufigkeit, bislang ohne neuen Flüssigkeitsaufbau.
Was das für die Zukunft bedeutet
Für Familien und Kliniker, die sich mit katastrophalen genetischen Epilepsien auseinandersetzen, zeigt diese Arbeit, dass das direkte Beruhigen eines überaktiven Ionenkanals mit einem maßgeschneiderten genetischen Medikament tatsächlich Anfälle reduzieren kann, selbst bei Kindern, die auf keine konventionelle Medikation angesprochen haben. Gleichzeitig unterstreicht das Auftreten von Hydrozephalus bei beiden behandelten Patienten, dass solche kraftvollen Therapien ernsthafte, noch unzureichend verstandene Risiken bergen können. Die Autoren schließen, dass die Hemmung des KCNT1-Kanals eine valide und vielversprechende Strategie ist, dass die Dosierung jedoch schrittweise erfolgen und zukünftige Studien sorgfältige Bildgebung des Gehirns sowie klinische Überwachung einschließen müssen. Breiter betrachtet hebt die Studie sowohl das transformativen Potenzial als auch die ethische Verantwortung hervor, die mit der Entwicklung individualisierter Genmedikamente für einige der schwerstkranken Patienten einhergeht.
Zitation: Nakayama, T., El Achkar, C.M., Burbano, L.E. et al. Antisense oligonucleotide-mediated knockdown therapy in two infants with severe KCNT1 epileptic encephalopathy. Nat Med 32, 1411–1420 (2026). https://doi.org/10.1038/s41591-026-04314-9
Schlüsselwörter: KCNT1-Epilepsie, Antisense-Oligonukleotid, Präzisionsmedizin, Säuglingskrämpfe, Hydrozephalus