Clear Sky Science · zh
KIF11通过抑制磷酸化驱动的PRDX1相分离阻止家族性渗出性玻璃体视网膜病变中视网膜内皮细胞的铁死亡
为何年幼眼睛中的脆弱血管重要
一些婴儿和幼儿会出现一种罕见的眼病,称为家族性渗出性玻璃体视网膜病变(FEVR),其特征是眼后方微小血管生长不良,视力可能因此遭受永久性损害。本研究提出了一个简单但关键的问题:构成这些血管内皮细胞的内部发生了什么故障,是否能找到一个可治疗的薄弱环节?研究者揭示了一条令人意外的事件链,牵涉到一种运动蛋白、一种保护性抗氧化酶,以及一种与铁和膜脂损伤相关的特殊细胞死亡形式。
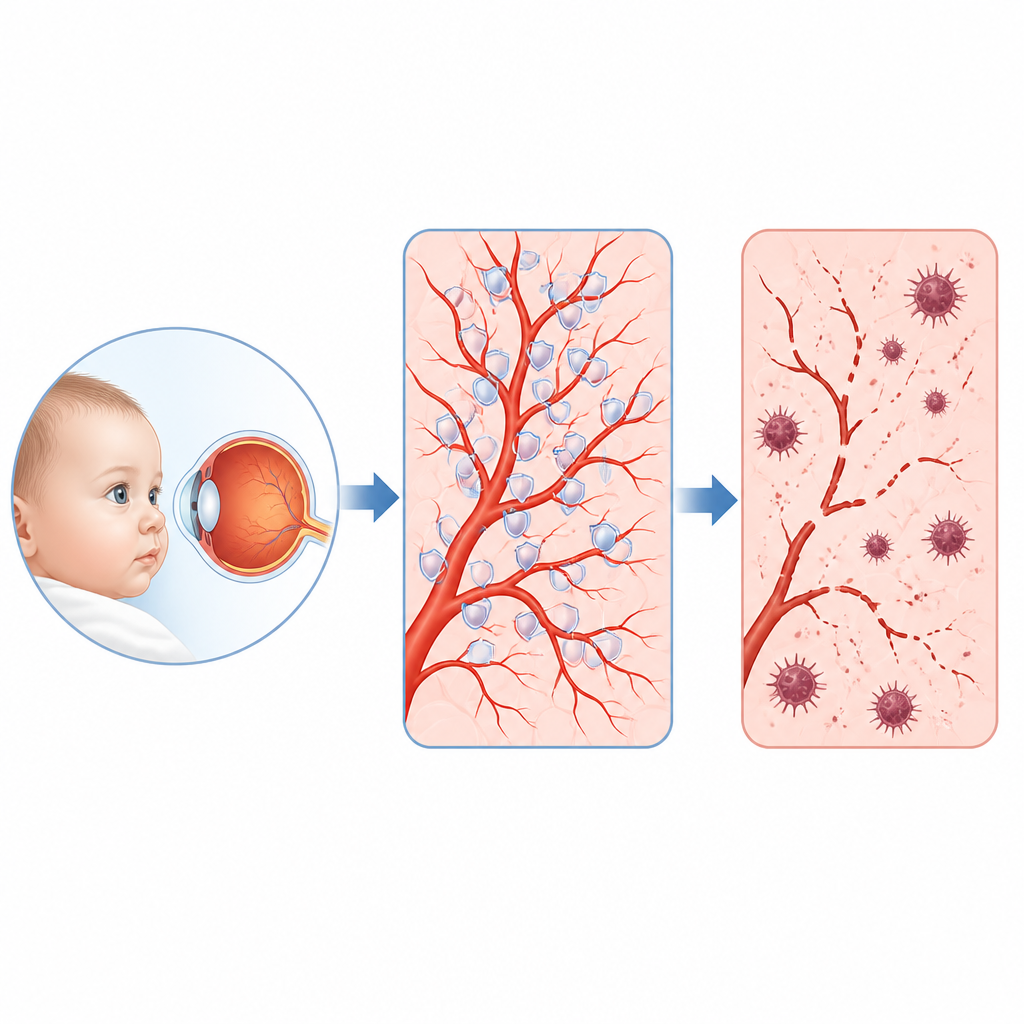
一种罕见儿童眼病及其隐秘的指挥链
临床上早已知道,许多患儿携带的基因缺陷会影响通常启动Norrin–β-连环蛋白通路的功能,而该通路指导视网膜血管的生长。但这些基因仅能解释约40%的病例,且它们在血管细胞中控制的关键靶点并不清楚。通过读取在人类视网膜血管细胞中该通路受损时哪些基因被开启或关闭,并分析来自小鼠视网膜的数千个单细胞,研究团队锁定了一个突出角色:称为KIF11的蛋白。KIF11曾主要被认为是一种协助细胞分裂的运动蛋白,但当Norrin–β-连环蛋白信号减弱时,KIF11表现为一个显著下调的枢纽分子。
从基因缺陷到血管细胞死亡
研究者接着测试了KIF11丧失时的后果。在培养皿中生长的人类视网膜血管细胞中,降低KIF11会减缓细胞增殖并导致细胞死亡。电子显微镜下,这些细胞显示出自噬结构增多和线粒体萎缩损伤的典型征象,这些特征与一种由铁驱动的细胞死亡——铁死亡相关。大规模的RNA和蛋白质测定证实,通常用于清除活性氧分子并防止细胞膜脂质氧化的保护系统被削弱,而氧化应激、铁积累和脂质过氧化的标志物上升。用一种阻断铁死亡的小分子处理这些细胞能够恢复许多缺陷,表明当KIF11缺失时,这一死亡程序是核心问题。
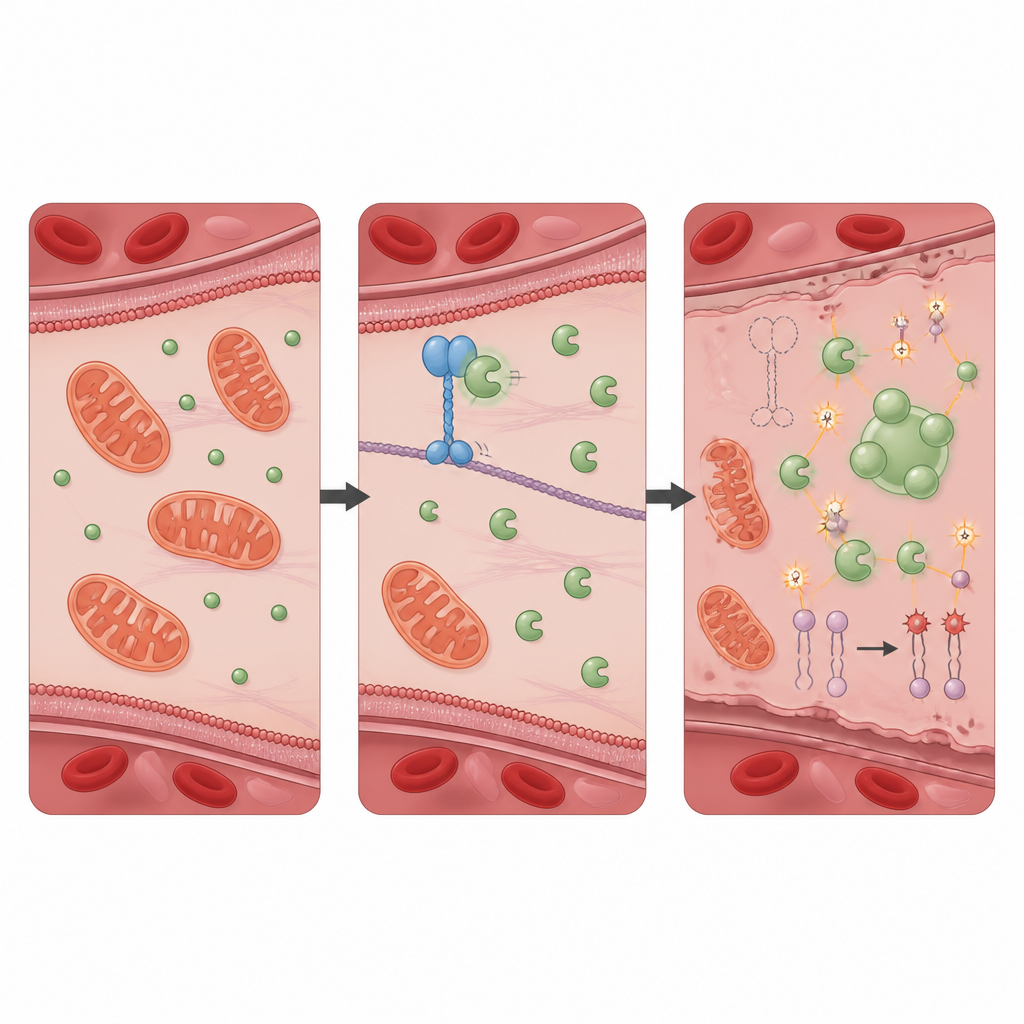
控制氧化损伤的保护性伙伴关系
为查明KIF11如何发挥保护作用,研究团队搜寻其物理相互作用的蛋白,鉴定出PRDX1——一种众所周知的抗氧化酶,可中和有害的过氧化物。在健康细胞中,KIF11与PRDX1结合,限制另一种蛋白Src对PRDX1特定位点的磷酸化。当KIF11缺失或因患者突变而截短时,Src更容易修饰PRDX1,促使这些酶分子聚集并在细胞内形成类液滴状的大团块。这种相分离降低了PRDX1的正常解毒活性,导致氧化应激上升。仅缺失PRDX1的细胞显示出几乎与缺失KIF11细胞相同的自噬增加、铁过载、膜损伤和铁死亡模式,而这些改变同样可被铁死亡抑制剂逆转。
从分子失效到动物体内的血管缺陷
小鼠模型帮助将这些微观事件与真实的血管缺陷联系起来。当β-连环蛋白信号仅在内皮细胞中关闭,或当Kif11在这些细胞中被删除时,新生小鼠的视网膜生长血管停滞且稀疏,近似模拟人类疾病。这些小鼠在血管丰富的组织中也显示出更多磷酸化的PRDX1、更多细胞自噬迹象,以及铁死亡相关蛋白的变化。在视网膜血管生长的短暂窗口期给予幼鼠铁死亡抑制剂,可部分恢复血管覆盖。同样地,输送额外的KIF11,或一种无法在关键位点被磷酸化、因此抵抗有害聚集的PRDX1变体,也能在没有明显副作用的情况下改善血管生长。
这对保护儿童视力意味着什么
总体而言,研究结果勾勒出一个明确的故事:控制视网膜血管生长的信号通路上调KIF11,而KIF11又维持抗氧化酶PRDX1的活性和均匀分布,防止失控的氧化损伤和铁驱动的细胞死亡在血管内皮细胞中发生。当遗传突变削弱这一通路时,KIF11水平下降,PRDX1被过度修饰并凝聚成液滴,铁死亡悄然消耗应负责构建与维护视网膜血管的细胞。尽管在将治疗应用于患者之前仍需更多研究,但该研究强调了两种可能有助于保护此病患儿视力的广义策略:直接靶向铁死亡,或稳定如PRDX1等抗氧化防御系统以保护发育中脆弱的眼睛。
引用: Yang, M., Zhao, R., Peng, L. et al. KIF11 prevents retinal endothelial ferroptosis in familial exudative vitreoretinopathy by inhibiting phosphorylation-driven PRDX1 phase separation. Nat Commun 17, 4360 (2026). https://doi.org/10.1038/s41467-026-71009-7
关键词: 视网膜血管, 铁死亡, 氧化应激, 家族性渗出性玻璃体视网膜病变, 抗氧化酶