Clear Sky Science · fr
KIF11 empêche la ferroptose des cellules endothéliales rétiniennes dans la rétinopathie exsudative familiale en inhibant la séparation de phase de PRDX1 induite par phosphorylation
Pourquoi les vaisseaux fragiles des yeux des jeunes comptent
Certs nourrissons et jeunes enfants développent une affection oculaire rare appelée rétinopathie exsudative familiale, dans laquelle les très petits vaisseaux à l’arrière de l’œil ne se développent pas correctement et la vision peut être endommagée de façon permanente. Cette étude pose une question simple mais cruciale : que se passe-t-il à l’intérieur des cellules qui tapissent ces vaisseaux, et peut-on identifier une faiblesse traitable ? Les chercheurs découvrent une chaîne d’événements surprenante impliquant une protéine motrice, une enzyme antioxydante protectrice et une forme particulière de mort cellulaire liée au fer et à l’altération des lipides membranaires.
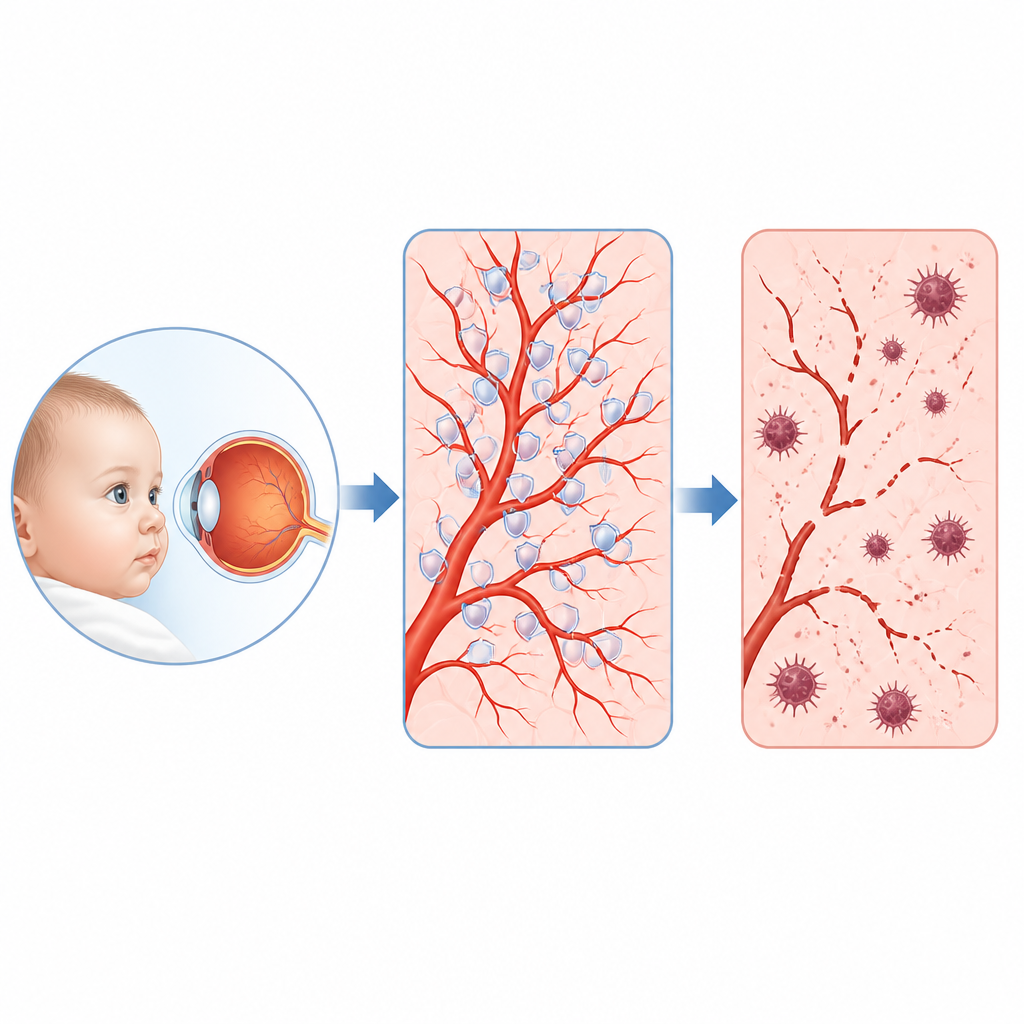
Une maladie oculaire infantile rare et sa hiérarchie cachée
Les cliniciens savent depuis longtemps que de nombreux enfants atteints présentent des anomalies dans des gènes qui activent normalement une voie appelée Norrin/bêta-caténine, laquelle guide la croissance des vaisseaux dans la rétine. Pourtant, ces gènes n’expliquent qu’environ 40 % des cas, et les cibles cruciales qu’ils contrôlent dans les cellules vasculaires restaient floues. En analysant les gènes dont l’expression change lorsque cette voie est perturbée dans des cellules vasculaires rétiniennes humaines, et en examinant des milliers de cellules uniques de rétines de souris, l’équipe a identifié un acteur remarquable : une protéine appelée KIF11. Connue autrefois principalement comme une protéine motrice participant à la division cellulaire, KIF11 s’est révélée être un centre majeur réprimé chaque fois que le signal Norrin/bêta-caténine était affaibli.
D’un gène défaillant à la mort des cellules vasculaires
Les chercheurs ont ensuite testé les conséquences de la perte de KIF11. Dans des cellules vasculaires rétiniennes humaines cultivées en laboratoire, la réduction de KIF11 ralentissait leur prolifération et entraînait la mort cellulaire. Au microscope électronique, ces cellules présentaient des signes caractéristiques d’augmentation des structures d’autonettoyage et des mitochondries endommagées et rétractées, un profil associé à un type de mort cellulaire ferroptotique dépendant du fer. Des mesures à grande échelle de l’ARN et des protéines ont confirmé que les systèmes protecteurs qui normalement détoxifient les molécules réactives de l’oxygène et empêchent l’oxydation des lipides membranaires étaient affaiblis, tandis que les marqueurs de stress oxydatif, d’accumulation de fer et de peroxydation lipidique augmentaient. Le traitement des cellules par une petite molécule bloquant la ferroptose a restauré bon nombre de ces anomalies, suggérant que ce programme de mort est un problème central en l’absence de KIF11.
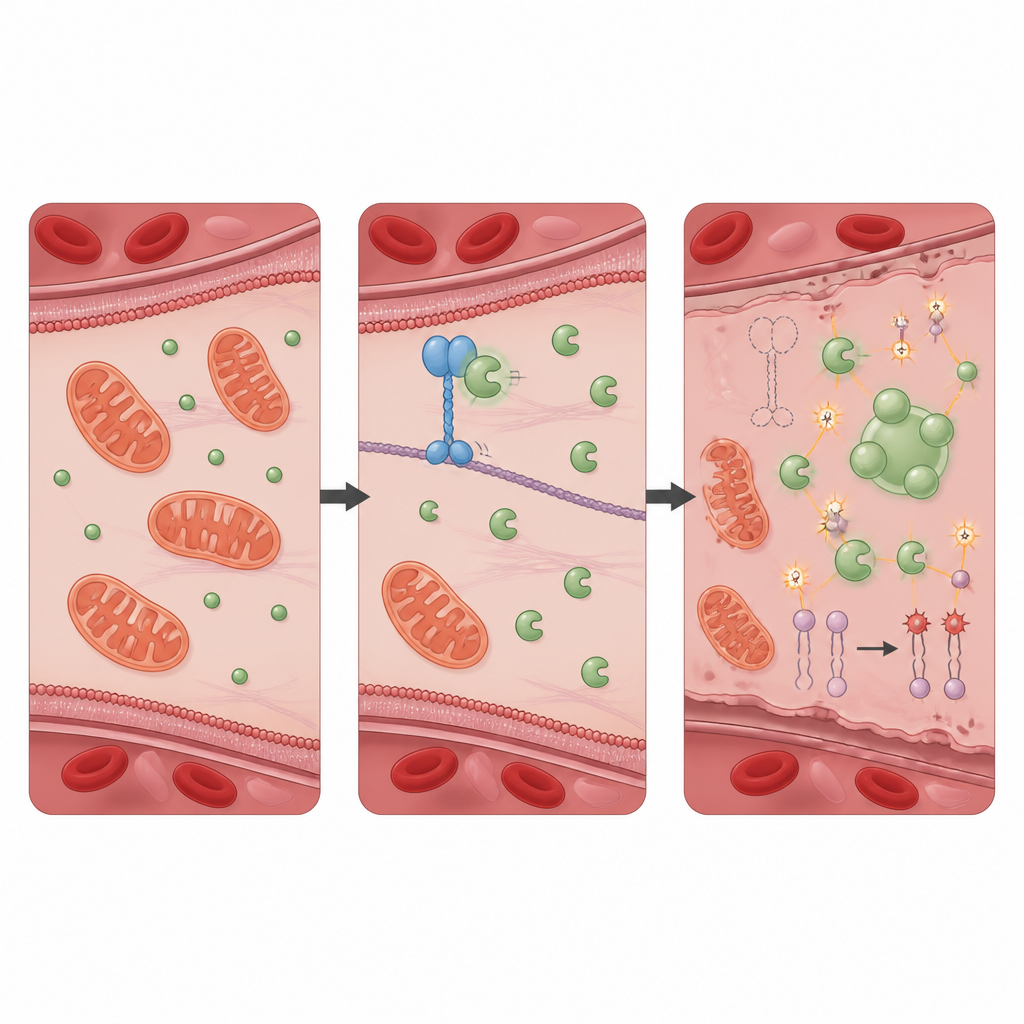
Un partenariat protecteur qui limite les dommages oxydatifs
Pour comprendre comment KIF11 exerce cet effet protecteur, l’équipe a recherché les protéines avec lesquelles elle interagit physiquement et a identifié PRDX1, une enzyme antioxydante bien connue qui neutralise les peroxydes nocifs. Dans les cellules saines, KIF11 se lie à PRDX1 et limite l’accès d’une autre protéine, Src, qui ajoute un groupe phosphate à PRDX1 en un site spécifique. Lorsque KIF11 est absent ou tronqué par des mutations observées chez des patients, Src peut plus facilement modifier PRDX1, incitant les molécules de l’enzyme à s’assembler en agrégats et à former des gouttelettes de type liquide à l’intérieur de la cellule. Cette séparation de phase réduit l’activité détoxifiante normale de PRDX1, permettant au stress oxydatif de s’accroître. Les cellules dépourvues uniquement de PRDX1 ont montré un schéma presque identique d’autodigestion, de surcharge en fer, de dommages membranaires et de mort ferroptotique que les cellules privées de KIF11, et ces changements étaient également inversés par l’inhibiteur de la ferroptose.
De la défaillance moléculaire aux vaisseaux défectueux chez l’animal
Des modèles murins ont permis de relier ces événements microscopiques à des défauts vasculaires réels. Lorsque le signal bêta-caténine était coupé uniquement dans les cellules endothéliales, ou lorsque Kif11 lui-même était supprimé dans ces cellules, les vaisseaux rétiniens en croissance chez les nouveau-nés mice s’arrêtaient et demeuraient clairsemés, reproduisant fidèlement la maladie humaine. Ces mêmes souris présentaient davantage de PRDX1 phosphorylée, plus de signes d’autodigestion cellulaire et des modifications des protéines liées à la ferroptose dans les tissus riches en vaisseaux. L’administration d’un inhibiteur de la ferroptose aux jeunes souris pendant la brève fenêtre de croissance des vaisseaux rétiniens a partiellement restauré la couverture vasculaire. De même, l’apport supplémentaire de KIF11, ou d’une version de PRDX1 qui ne peut pas être phosphorylée au site critique et résiste ainsi à l’agrégation néfaste, a amélioré la croissance vasculaire sans effets secondaires évidents.
Ce que cela signifie pour la protection de la vision chez l’enfant
Ensemble, ces résultats dessinent un récit clair : une voie de signalisation qui contrôle la croissance des vaisseaux rétiniens stimule KIF11, qui à son tour maintient PRDX1 actif et distribué de manière homogène, empêchant une montée incontrôlée des dommages oxydatifs et la mort cellulaire induite par le fer dans les cellules endothéliales. Lorsque cette voie est affaiblie par des mutations héréditaires, les niveaux de KIF11 chutent, PRDX1 est excessivement modifié et s’agglutine en gouttelettes, et la ferroptose érode silencieusement les cellules qui devraient construire et entretenir la vascularisation rétinienne. Bien que des travaux supplémentaires soient nécessaires avant d’aboutir à des traitements pour les patients, l’étude met en lumière deux stratégies générales susceptibles d’aider à protéger la vision dans cette maladie : cibler la ferroptose elle-même, ou stabiliser les défenses antioxydantes comme PRDX1 dans l’œil en développement et fragile.
Citation: Yang, M., Zhao, R., Peng, L. et al. KIF11 prevents retinal endothelial ferroptosis in familial exudative vitreoretinopathy by inhibiting phosphorylation-driven PRDX1 phase separation. Nat Commun 17, 4360 (2026). https://doi.org/10.1038/s41467-026-71009-7
Mots-clés: vaisseaux sanguins rétiniens, ferroptose, stress oxydatif, rétinopathie exsudative familiale, enzymes antioxydantes