Clear Sky Science · de
KIF11 verhindert Ferroptose retinaler Endothelzellen bei familiärer exsudativer Vitreoretinopathie durch Hemmung der phosphorylierungsgetriebenen Phasentrennung von PRDX1
Warum fragile Blutgefäße in jungen Augen wichtig sind
Einige Säuglinge und Kleinkinder entwickeln eine seltene Augenerkrankung namens familiäre exsudative Vitreoretinopathie, bei der die feinen Blutgefäße im hinteren Teil des Auges nicht richtig ausreifen und das Sehvermögen dauerhaft geschädigt werden kann. Die Studie stellt eine einfache, aber entscheidende Frage: Was läuft in den Zellen, die diese Gefäße auskleiden, schief, und lässt sich eine verwundbare Stelle finden, die sich behandeln ließe? Die Forschenden decken eine überraschende Kette von Ereignissen auf, an der ein Motorprotein, ein schützendes antioxidatives Enzym und eine spezielle Form des Zelltods beteiligt sind, die mit Eisen und Lipidschäden in Zellmembranen verbunden ist.
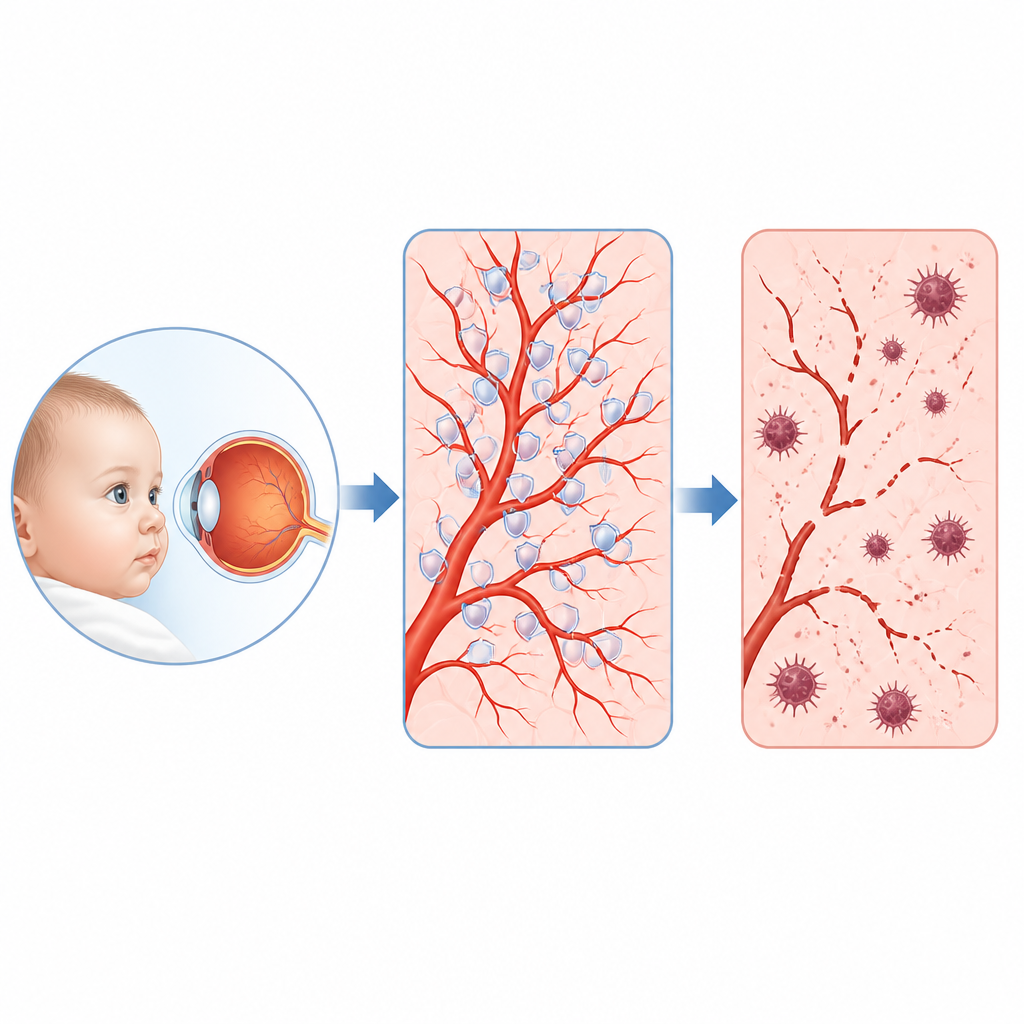
Eine seltene kindliche Augenerkrankung und ihre verborgene Kommandokette
Ärztinnen und Ärzte wissen seit Langem, dass viele Kinder mit dieser Erkrankung Veränderungen in Genen tragen, die normalerweise einen Signalweg namens Norrin‑/β‑Catenin aktivieren, der das Wachstum retinaler Blutgefäße steuert. Diese Gene erklären jedoch nur etwa 40 Prozent der Fälle, und die entscheidenden Zielproteine in den Gefäßzellen blieben unklar. Durch das Auslesen, welche Gene an‑ oder abgeschaltet werden, wenn dieser Signalweg in menschlichen retinalen Gefäßzellen gestört ist, und durch die Analyse tausender Einzelzellen aus Mausretinas, konnte das Team einen herausragenden Akteur identifizieren: das Protein KIF11. Früher vor allem als Motorprotein bekannt, das bei der Zellteilung hilft, erwies sich KIF11 als wichtiges Zentrum, das herunterreguliert wird, sobald das Norrin‑/β‑Catenin‑Signal geschwächt ist.
Vom fehlerhaften Gen zu sterbenden Gefäßzellen
Anschließend prüften die Forschenden, was passiert, wenn KIF11 verloren geht. In humanen retinalen Gefäßzellen in Kultur verlangsamte die Reduktion von KIF11 deren Proliferation und führte zu Zellsterben. Im Elektronenmikroskop zeigten diese Zellen typische Zeichen vermehrter Autophagie und geschädigter, verkümmerter Mitochondrien – ein Muster, das mit einer Eisen‑vermittelten Form des Zelltods namens Ferroptose in Verbindung gebracht wird. Großmaßstäbliche Messungen von RNA und Proteinen bestätigten, dass Schutzsysteme, die normalerweise reaktive Sauerstoffspezies entgiften und ein Ranzigwerden von Membranlipiden verhindern, geschwächt waren, während Marker für oxidativen Stress, Eisenansammlung und Lipidperoxidation zunahmen. Die Behandlung der Zellen mit einer kleinen Verbindung, die Ferroptose blockiert, stellte viele dieser Defekte wieder her, was darauf hindeutet, dass dieses Todesprogramm ein zentrales Problem bei KIF11‑Mangel ist.
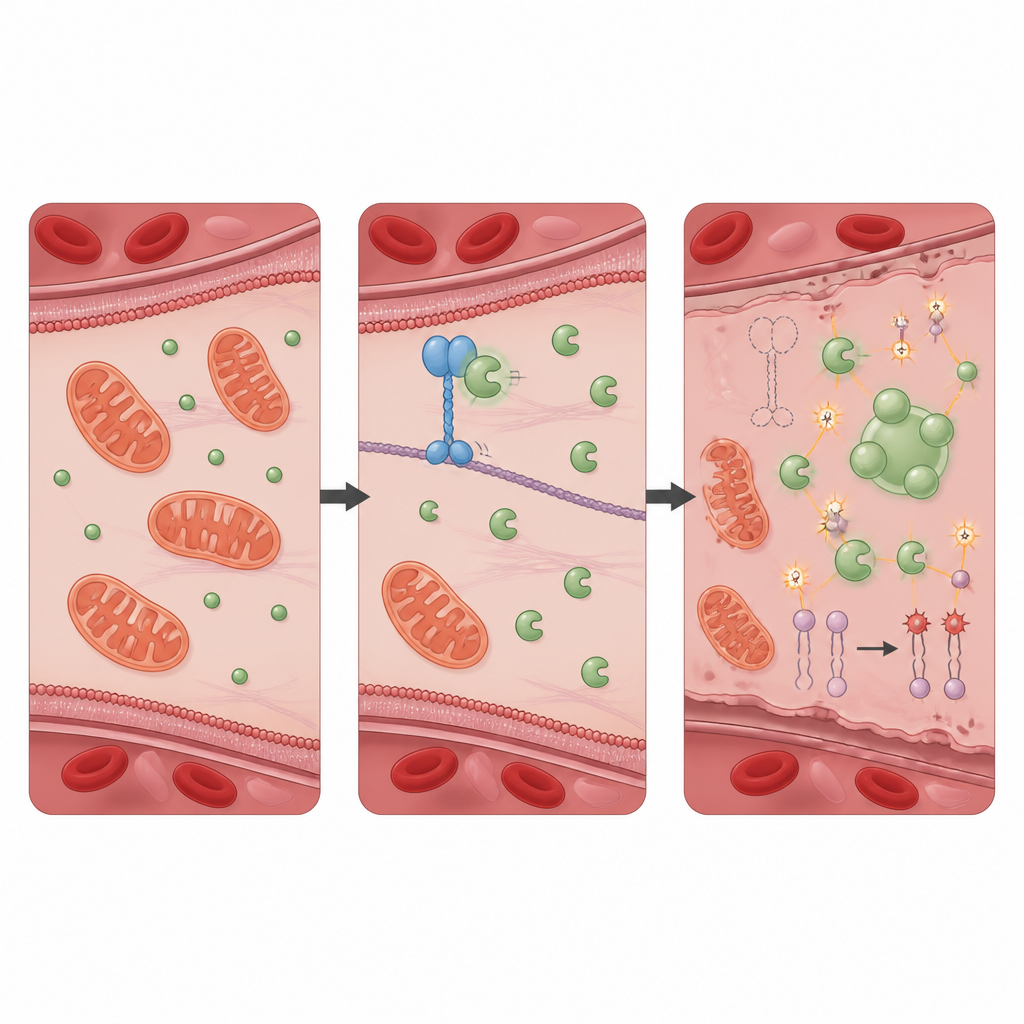
Eine schützende Partnerschaft, die oxidative Schäden in Schach hält
Um herauszufinden, wie KIF11 diesen Schutz vermittelt, suchte das Team nach Proteinen, mit denen es physisch interagiert, und identifizierte PRDX1, ein bekanntes antioxidatives Enzym, das schädliche Peroxide neutralisiert. In gesunden Zellen bindet KIF11 PRDX1 und begrenzt den Zugang eines anderen Proteins, Src, das eine Phosphatgruppe an einer bestimmten Stelle von PRDX1 anbringt. Fehlt KIF11 oder ist es durch Patientenmutationen verkürzt, kann Src PRDX1 leichter modifizieren, wodurch die Enzymmoleküle dazu neigen, zu größeren Clustern zusammenzutreten und im Zellinneren flüssigkeitsähnliche Tröpfchen zu bilden. Diese Phasentrennung reduziert die normale entgiftende Aktivität von PRDX1 und lässt den oxidativen Stress ansteigen. Zellen, denen allein PRDX1 fehlte, zeigten ein nahezu identisches Muster aus Autophagie, Eisenüberladung, Membranschäden und ferroptotischem Zelltod wie Zellen ohne KIF11; auch diese Änderungen ließen sich durch den Ferroptose‑Inhibitor rückgängig machen.
Vom molekularen Versagen zu fehlerhaften Blutgefäßen im Tiermodell
Mausmodelle halfen, diese mikroskopischen Ereignisse mit realen Gefäßdefekten zu verknüpfen. Wenn das β‑Catenin‑Signal ausschließlich in Endothelzellen abgeschaltet wurde oder wenn Kif11 in diesen Zellen gelöscht wurde, blieben die wachsenden retinalen Gefäße neugeborener Mäuse stehen und waren spärlich – ein klares Abbild der humanen Erkrankung. Dieselben Mäuse zeigten vermehrt phosphoryliertes PRDX1, stärkere Zeichen zellulärer Selbstverdauung und Veränderungen ferroptosebezogener Proteine in gefäßreichen Geweben. Die Gabe eines Ferroptose‑Inhibitors an junge Mäuse während des kurzen Zeitfensters des retinalen Gefäßwachstums stellte die Gefäßbedeckung teilweise wieder her. Ebenso verbesserten die Zuführung von zusätzlichem KIF11 oder einer PRDX1‑Variante, die an der kritischen Stelle nicht phosphorylierbar ist und somit schädlichem Zusammenlagern widersteht, das Gefäßwachstum ohne offensichtliche Nebenwirkungen.
Was das für den Schutz des Sehvermögens bei Kindern bedeutet
In der Summe zeichnen die Befunde eine klare Geschichte: Ein Signalweg, der das Wachstum retinaler Gefäße steuert, fördert KIF11, das seinerseits das antioxidative PRDX1 aktiv und gleichmäßig verteilt hält und so eskalierende oxidative Schäden und eisengetriebenen Zelltod in den Endothelzellen verhindert. Fällt dieser Weg durch vererbte Mutationen schwächer aus, sinken die KIF11‑Spiegel, wird PRDX1 übermäßig modifiziert und verklumpt zu Tröpfchen, und Ferroptose zermürbt stillschweigend die Zellen, die die Netzhautgefäße aufbauen und erhalten sollten. Zwar sind weitere Studien nötig, bevor Therapien Patienten erreichen, doch hebt die Arbeit zwei weit gefasste Strategien hervor, die das Sehvermögen bei dieser Erkrankung schützen könnten: das gezielte Hemmen von Ferroptose oder die Stabilisierung antioxidativer Abwehrmechanismen wie PRDX1 im verletzlichen sich entwickelnden Auge.
Zitation: Yang, M., Zhao, R., Peng, L. et al. KIF11 prevents retinal endothelial ferroptosis in familial exudative vitreoretinopathy by inhibiting phosphorylation-driven PRDX1 phase separation. Nat Commun 17, 4360 (2026). https://doi.org/10.1038/s41467-026-71009-7
Schlüsselwörter: retinale Blutgefäße, Ferroptose, oxidativer Stress, familiäre exsudative Vitreoretinopathie, antioxidative Enzyme