Clear Sky Science · pl
KIF11 zapobiega ferroptozie śródbłonka siatkówki w rodzinnym wysiękowym odwarstwieniu ciała szklistego poprzez hamowanie fosforylacji napędzającej separację fazową PRDX1
Dlaczego kruche naczynia u młodych oczu mają znaczenie
U niektórych niemowląt i małych dzieci rozwija się rzadkie schorzenie oka zwane rodzinnym wysiękowym odwarstwieniem ciała szklistego, w którym drobne naczynia krwionośne w tylnej części oka nie rozwijają się prawidłowo, a wzrok może ulec trwałemu uszkodzeniu. W tym badaniu postawiono proste, lecz kluczowe pytanie: co zawodzi wewnątrz komórek wyściełających te naczynia i czy można znaleźć słaby punkt, który da się leczyć? Naukowcy odkryli zaskakujący łańcuch zdarzeń obejmujący białko motoryczne, ochronny enzym antyoksydacyjny i szczególną formę śmierci komórkowej związaną z żelazem i uszkodzeniem lipidów błonowych.
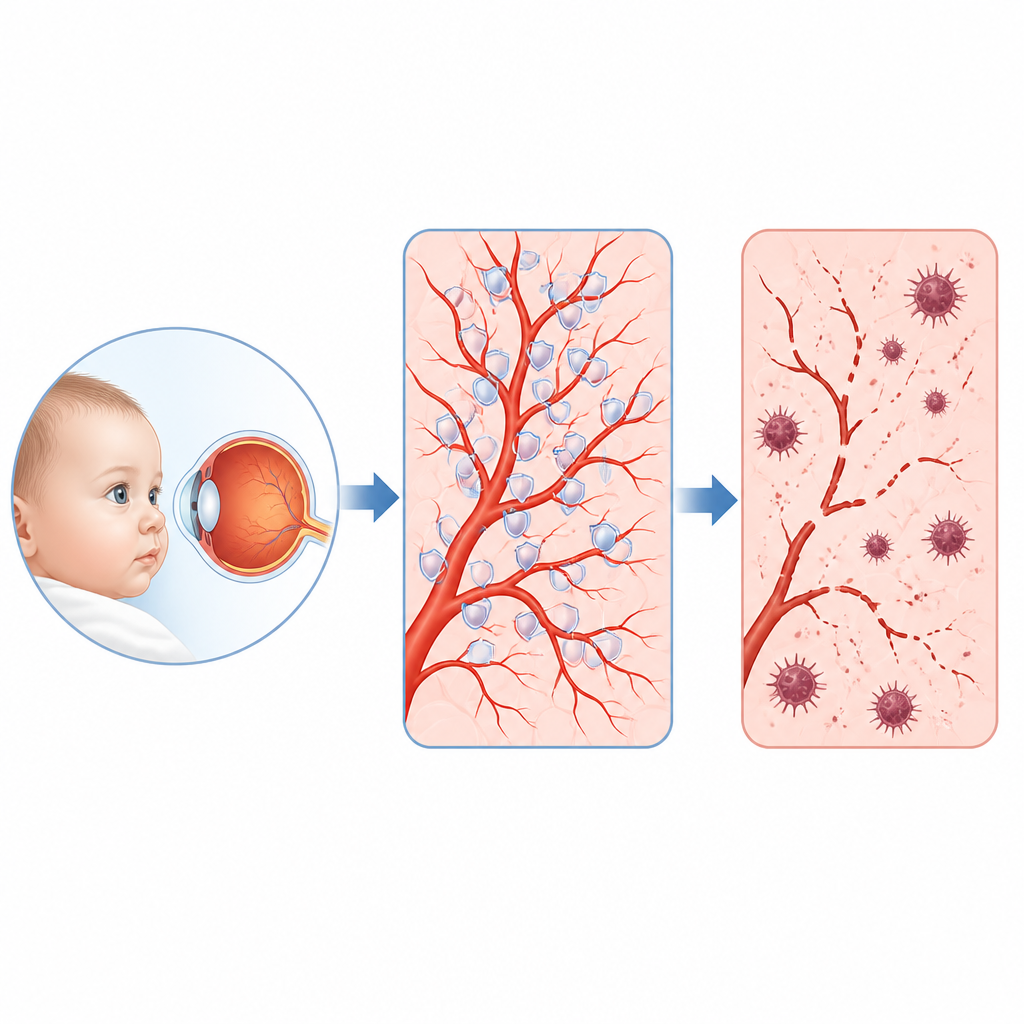
Rzadkie dziecięce schorzenie oka i jego skryty łańcuch dowodzenia
Lekarze od dawna wiedzą, że u wielu dzieci z tym schorzeniem występują wady w genach, które normalnie włączają szlak zwany Norrin–beta-katenina, kierujący wzrostem naczyń w siatkówce. Jednak te geny tłumaczą tylko około 40 procent przypadków, a kluczowe cele ich działania w komórkach naczyń pozostawały niejasne. Czytając, które geny są włączane lub wyłączane, gdy ten szlak jest zaburzony w ludzkich komórkach naczyń siatkówki, oraz analizując tysiące pojedynczych komórek z mysich siatkówek, zespół wytypował jednego wyraźnego uczestnika: białko o nazwie KIF11. Kiedyś znane głównie jako motor pomagający w podziale komórek, KIF11 okazało się istotnym węzłem, który był tłumiony za każdym razem, gdy sygnał Norrin–beta-katenina był osłabiony.
Od wadliwego genu do ginących komórek naczyń
Następnie badacze sprawdzili, co się dzieje po utracie KIF11. W hodowlach ludzkich komórek naczyń siatkówki zmniejszenie poziomu KIF11 spowalniało ich wzrost i prowadziło do śmierci komórek. W mikroskopie elektronowym komórki te wykazywały charakterystyczne objawy zwiększonej aktywności autofagii i uszkodzonych, skurczonych mitochondriów — wzorzec powiązany z rodzajem śmierci komórkowej napędzanej przez żelazo, zwanej ferroptozą. Pomiary na dużą skalę RNA i białek potwierdziły, że systemy ochronne zwykle detoksykujące reaktywne formy tlenu i zapobiegające utlenianiu lipidów w błonach komórkowych zostały osłabione, podczas gdy markery stresu oksydacyjnego, gromadzenia żelaza i peroksydacji lipidów wzrosły. Leczenie komórek małą cząsteczką blokującą ferroptozę przywróciło wiele z tych defektów, co sugeruje, że ten program śmierci jest centralnym problemem przy braku KIF11.
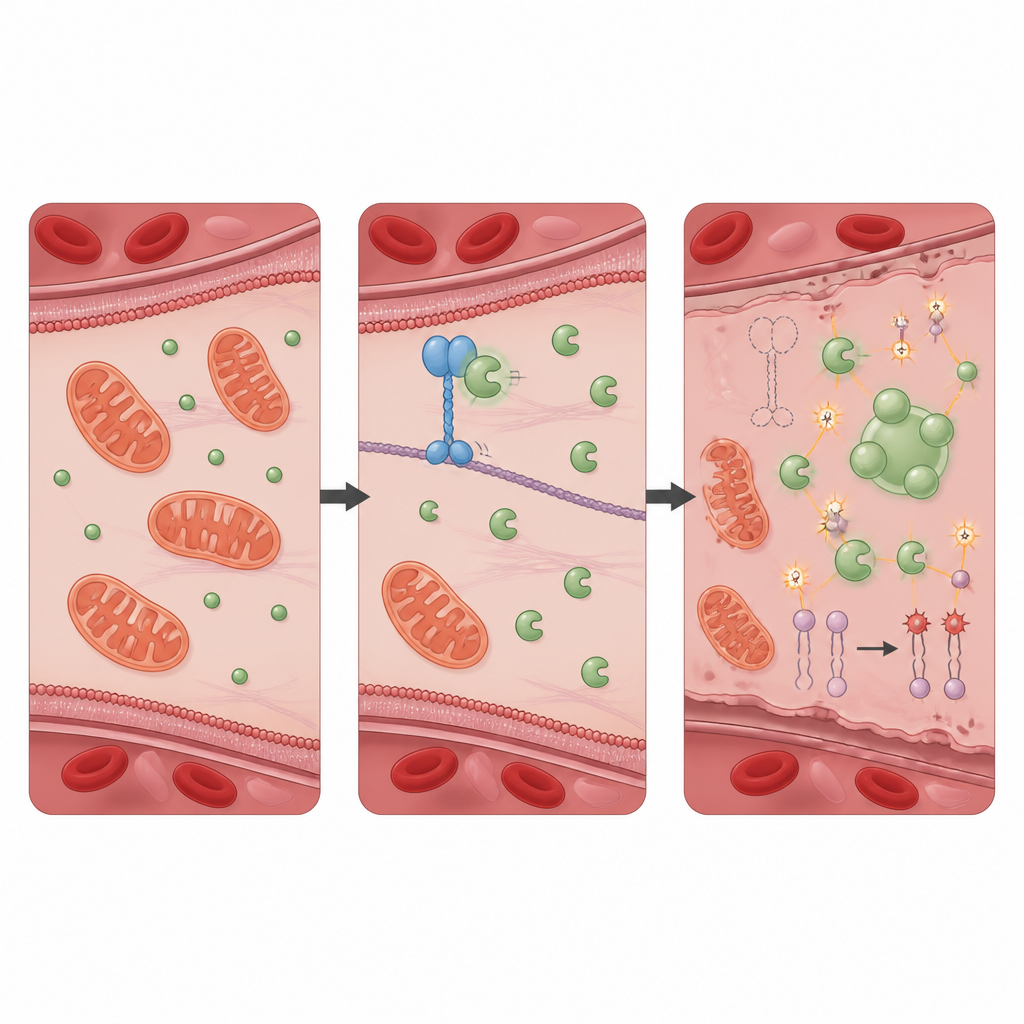
Ochronne partnerstwo, które opanowuje uszkodzenia oksydacyjne
Aby dowiedzieć się, jak KIF11 wywiera ten efekt ochronny, zespół poszukał białek, z którymi fizycznie oddziałuje, i zidentyfikował PRDX1, dobrze znany enzym antyoksydacyjny neutralizujący szkodliwe nadtlenki. W zdrowych komórkach KIF11 wiąże się z PRDX1 i ogranicza dostęp innego białka, Src, które dodaje grupę fosforanową do PRDX1 w specyficznym miejscu. Gdy KIF11 jest nieobecny lub skrócony przez mutacje pacjentów, Src łatwiej modyfikuje PRDX1, co skłania cząsteczki enzymu do łączenia się w większe skupiska i tworzenia płynopodobnych kropli wewnątrz komórki. Ta separacja fazowa zmniejsza normalną aktywność detoksykacyjną PRDX1, umożliwiając wzrost stresu oksydacyjnego. Komórki pozbawione samego PRDX1 wykazywały niemal identyczny wzorzec autofagii, przeładowania żelazem, uszkodzeń błon i śmierci ferroptotycznej jak komórki bez KIF11, a zmiany te także odwrócono przez inhibitor ferroptozy.
Od molekularnej awarii do wadliwych naczyń u zwierząt
Modele mysie pomogły powiązać te mikroskopowe zdarzenia z rzeczywistymi defektami naczyń. Gdy sygnał beta-kateniny był wyłączony tylko w komórkach śródbłonka, lub gdy sam Kif11 był usunięty w tych komórkach, rosnące naczynia siatkówki u nowo narodzonych myszy zatrzymywały się i były skąpe, co wiernie naśladowało ludzką chorobę. Te same myszy wykazywały więcej sfosforylowanego PRDX1, więcej oznak autofagii oraz przesunięć w białkach związanych z ferroptozą w tkankach bogatych w naczynia krwionośne. Podanie młodym myszom inhibitora ferroptozy podczas krótkiego okna wzrostu naczyń siatkówki częściowo przywróciło pokrycie naczyń. Podobnie dostarczenie dodatkowego KIF11 lub wersji PRDX1, której nie można sfosforylować w krytycznym miejscu i która odporna jest na szkodliwe skupianie, poprawiło wzrost naczyń bez oczywistych skutków ubocznych.
Co to oznacza dla ochrony wzroku u dzieci
Razem wyniki przedstawiają jasną historię: szlak sygnałowy kontrolujący wzrost naczyń siatkówki zwiększa poziom KIF11, który z kolei utrzymuje PRDX1 aktywny i równomiernie rozmieszczony, zapobiegając wymykającemu się spod kontroli uszkodzeniu oksydacyjnemu i śmierci komórek napędzanej przez żelazo w komórkach wyściełających naczynia. Gdy ten szlak jest osłabiony przez dziedziczne mutacje, poziomy KIF11 spadają, PRDX1 ulega nadmiernej modyfikacji i zlepia się w krople, a ferroptoza cicho niszczy komórki, które powinny budować i utrzymywać unaczynienie siatkówki. Choć potrzeba jeszcze badań, zanim terapie trafią do pacjentów, badanie wskazuje dwie szerokie strategie, które mogą pomóc chronić wzrok w tym schorzeniu: ukierunkowanie na samą ferroptozę lub stabilizowanie mechanizmów antyoksydacyjnych, takich jak PRDX1, w kruchej rozwijającej się siatkówce.
Cytowanie: Yang, M., Zhao, R., Peng, L. et al. KIF11 prevents retinal endothelial ferroptosis in familial exudative vitreoretinopathy by inhibiting phosphorylation-driven PRDX1 phase separation. Nat Commun 17, 4360 (2026). https://doi.org/10.1038/s41467-026-71009-7
Słowa kluczowe: naczynia krwionośne siatkówki, ferroptoza, stres oksydacyjny, rodzinne wysiękowe odwarstwienie ciała szklistego, enzymy antyoksydacyjne