Clear Sky Science · sv
KIF11 förhindrar ferroptos i retinala endotelceller vid familjär exsudativ glaskroppsretinopati genom att hämma fosforyleringsdriven PRDX1-fasseparation
Varför sköra blodkärl i unga ögon spelar roll
Vissa spädbarn och småbarn utvecklar en sällsynt ögonsjukdom kallad familjär exsudativ glaskroppsretinopati, där de fina blodkärlen längst bak i ögat inte växer normalt och synen kan skadas permanent. Denna studie ställer en enkel men avgörande fråga: vad går fel inne i cellerna som bekläder dessa kärl, och kan vi hitta en svag punkt som kan behandlas? Forskarna avslöjar en överraskande händelsekedja som involverar ett motorprotein, ett skyddande antioxidantenzym och en särskild form av celldöd kopplad till järn och fettskador i cellmembran.
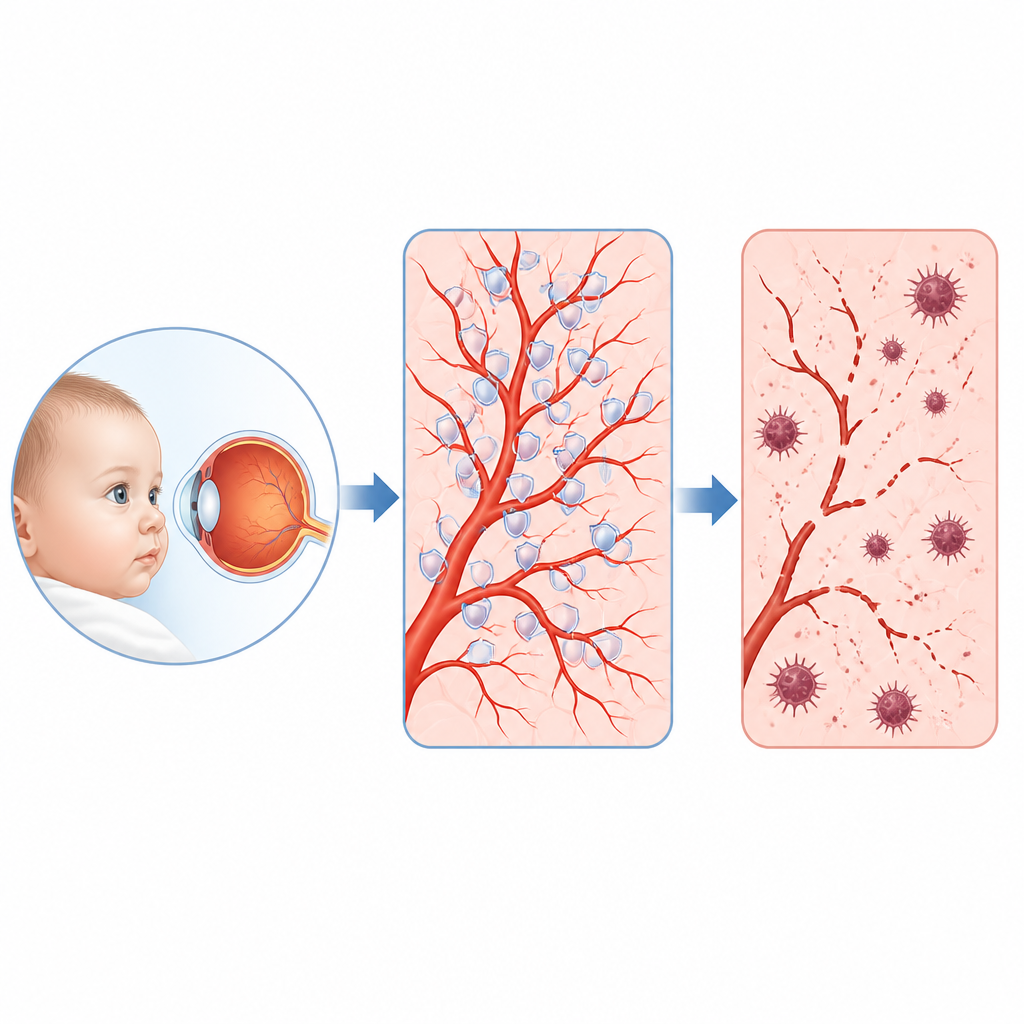
En sällsynt barndomssjukdom och dess dolda kommandokedja
Läkare har länge vetat att många barn med denna sjukdom bär fel i gener som normalt aktiverar en signalväg kallad Norrin–beta‑catenin, vilken styr blodkärlstillväxt i näthinnan. Ändå förklarar dessa gener bara cirka 40 procent av fallen, och de avgörande målen de reglerar i kärlcellerna var tidigare oklara. Genom att läsa av vilka gener som slås på eller av när denna väg störs i mänskliga retinala kärlceller, och genom att analysera tusentals enskilda celler från musnäthinnor, identifierade teamet en framträdande aktör: ett protein kallat KIF11. Tidigare mest känt som en motor som hjälper celler att dela sig, visade sig KIF11 vara en viktig nod som nedregleras när Norrin–beta‑catenin‑signalen försvagas.
Från felaktig gen till döende kärlceller
Nästa steg för forskarna var att testa vad som händer när KIF11 förloras. I humana retinala kärlceller odlade i petriskålar ledde reducerad KIF11 till saktare tillväxt och cellers död. I elektronmikroskop visade dessa celler typiska tecken på ökade autofagi‑strukturer och skadade, förskrympta mitokondrier — ett mönster kopplat till en järndriven celldödstyp kallad ferroptos. Storskaliga mätningar av RNA och proteiner bekräftade att skyddssystem som normalt avgiftar reaktiva syremolekyler och förhindrar att lipider i cellmembranen oxiderar försvagades, medan markörer för oxidativ stress, järnansamling och lipidsyraoxidation ökade. Behandling av cellerna med ett litet molekylärt ämne som blockerar ferroptos återställde många av dessa defekter, vilket tyder på att detta dödsprogram är ett centralt problem när KIF11 saknas.
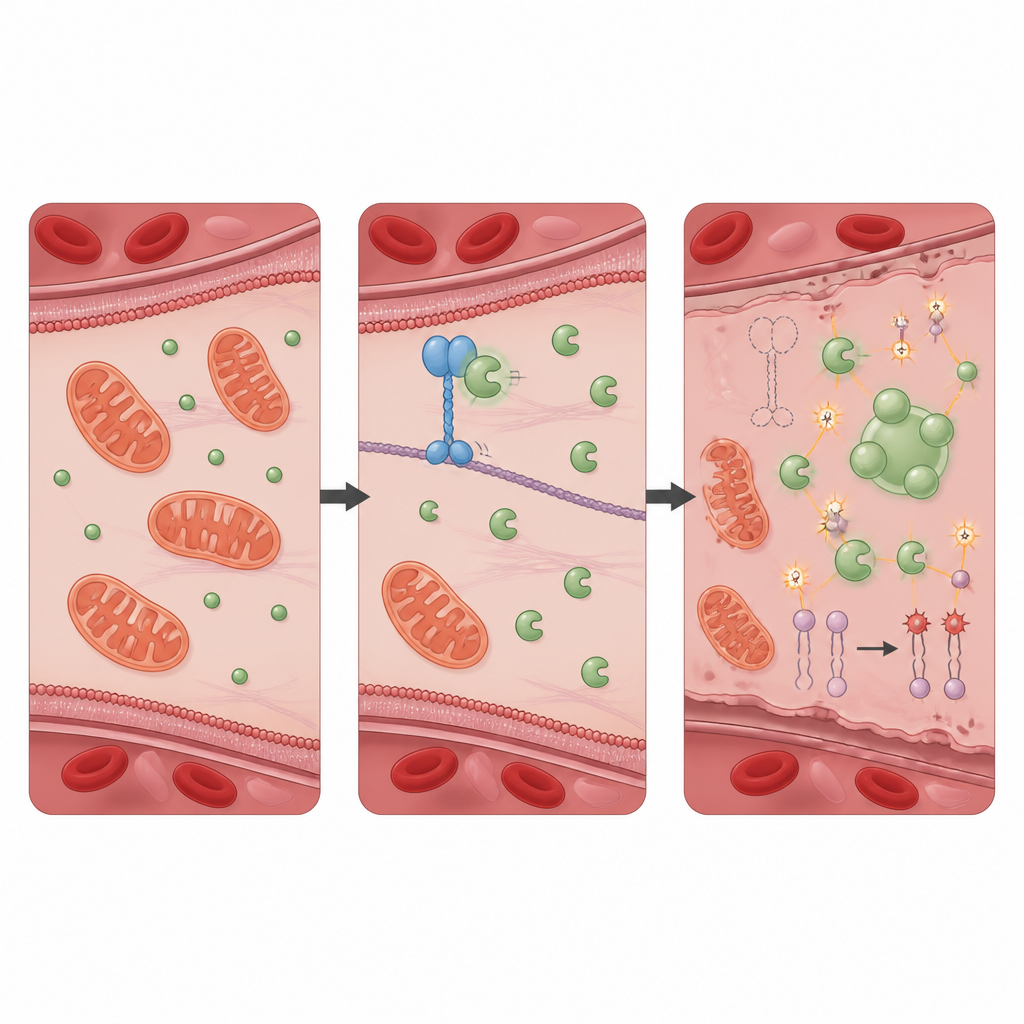
Ett skyddande partnerskap som håller oxidativ skada i schack
För att ta reda på hur KIF11 utövar denna skyddande effekt sökte teamet efter proteiner som det fysiskt interagerar med och identifierade PRDX1, ett välkänt antioxidantenzym som neutraliserar skadliga peroxider. I friska celler binder KIF11 PRDX1 och begränsar tillgången för ett annat protein, Src, som tillsätter en fosfatgrupp på PRDX1 vid en specifik plats. När KIF11 saknas eller förkortas av patientmutationer kan Src lättare modifiera PRDX1, vilket får enzymmolekylerna att gå ihop i större kluster och bilda vätskeliknande droppar inne i cellen. Denna fässeparation minskar PRDX1:s normala avgiftande aktivitet och låter den oxidativa stressen öka. Celler som saknar enbart PRDX1 visade ett nästan identiskt mönster av autofagi, järnöverbelastning, membranskada och ferroptotisk död som celler utan KIF11, och dessa förändringar kunde också återställas av ferroptos‑hämmaren.
Från molekylärt fel till defekta blodkärl i djur
Musmodeller hjälpte till att koppla dessa mikroskopiska händelser till verkliga kärldefekter. När beta‑catenin‑signalen stängdes av endast i endotelceller, eller när Kif11 själv togs bort i dessa celler, stannade de växande retinala kärlen hos nyfödda möss av och blev glesa — en bild som starkt liknar den mänskliga sjukdomen. Samma möss visade mer fosforylerad PRDX1, fler tecken på cellulär autofagi och förskjutningar i ferroptos‑relaterade proteiner i vävnader rika på blodkärl. Att ge unga möss en ferroptos‑inhibitor under det korta fönstret för retinal kärltillväxt återställde delvis kärltäckningen. På samma sätt förbättrade tillförsel av extra KIF11, eller en version av PRDX1 som inte kan fosforyleras vid den kritiska platsen och därmed motstår skadligt klustrande, kärltillväxten utan uppenbara biverkningar.
Vad detta betyder för att skydda synen hos barn
Tillsammans skisserar fynden en tydlig berättelse: en signalväg som kontrollerar retinal kärltillväxt uppreglerar KIF11, vilket i sin tur håller antioxidantenzymet PRDX1 aktivt och jämnt fördelat, och förhindrar okontrollerad oxidativ skada och järndriven celldöd i kärlendotelet. När denna väg försvagas av ärftliga mutationer faller KIF11‑nivåerna, PRDX1 övermodifieras och klumpar ihop sig till droppar, och ferroptos urholkar i det tysta de celler som ska bygga och underhålla näthinnans kärl. Mer arbete krävs innan behandlingar når patienter, men studien lyfter fram två breda strategier som kan hjälpa till att skydda synen vid detta tillstånd: att rikta in sig på själva ferroptosen eller att stabilisera antioxidantförsvar som PRDX1 i det känsliga utvecklande ögat.
Citering: Yang, M., Zhao, R., Peng, L. et al. KIF11 prevents retinal endothelial ferroptosis in familial exudative vitreoretinopathy by inhibiting phosphorylation-driven PRDX1 phase separation. Nat Commun 17, 4360 (2026). https://doi.org/10.1038/s41467-026-71009-7
Nyckelord: retinala blodkärl, ferroptos, oxidativ stress, familjär exsudativ glaskroppsretinopati, antioxidantenzym