Clear Sky Science · en
KIF11 prevents retinal endothelial ferroptosis in familial exudative vitreoretinopathy by inhibiting phosphorylation-driven PRDX1 phase separation
Why fragile blood vessels in young eyes matter
Some babies and young children develop a rare eye condition called familial exudative vitreoretinopathy, in which the tiny blood vessels at the back of the eye fail to grow properly and vision can be permanently damaged. This study asks a simple but crucial question: what goes wrong inside the cells lining those vessels, and can we find a weak link that might be treated? The researchers uncover a surprising chain of events involving a motor protein, a protective antioxidant enzyme, and a special form of cell death tied to iron and fat damage in cell membranes.
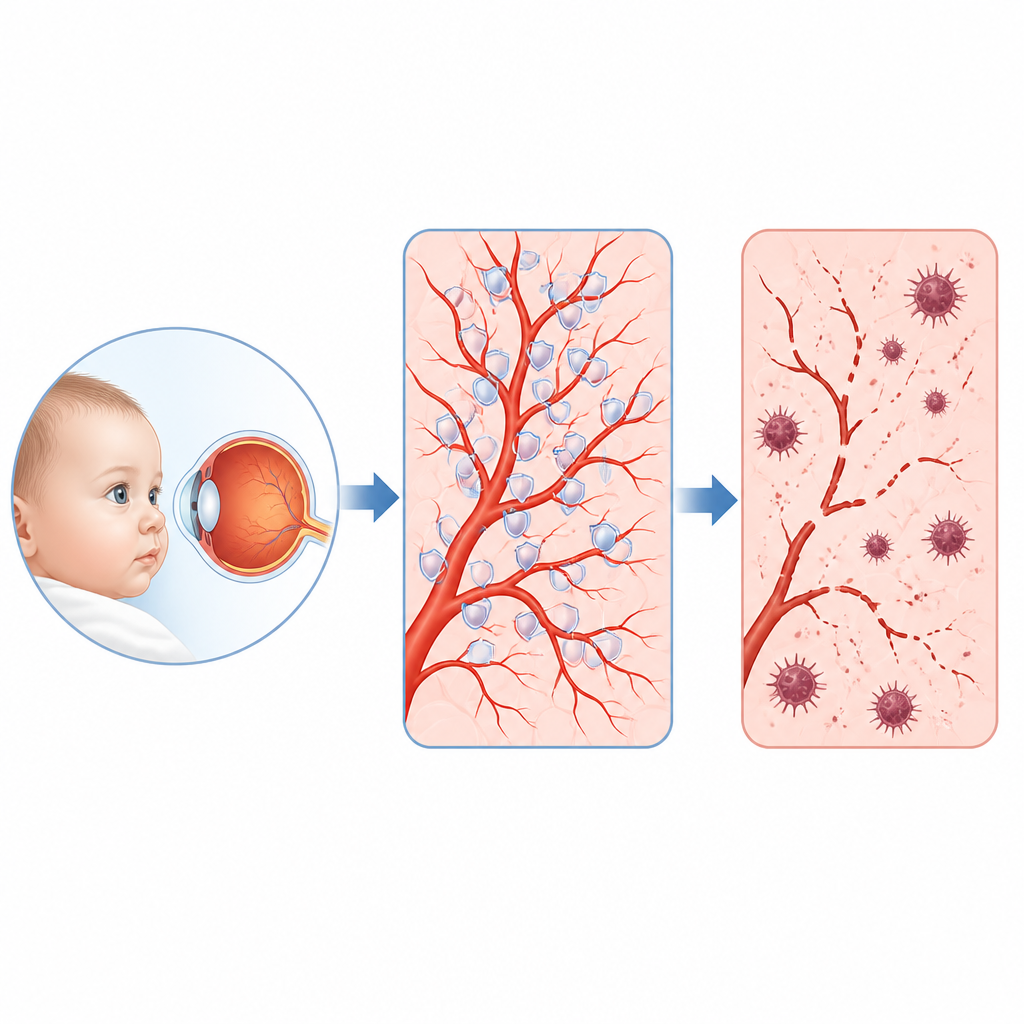
A rare childhood eye disease and its hidden chain of command
Doctors have long known that many children with this disease carry faults in genes that normally switch on a pathway called Norrin beta catenin, which guides blood vessel growth in the retina. Yet these genes explain only about 40 percent of cases, and the crucial targets they control in vessel cells were unclear. By reading out which genes switch on or off when this pathway is disrupted in human retinal vessel cells, and by analyzing thousands of single cells from mouse retinas, the team pinpointed one standout player: a protein called KIF11. Once known mainly as a motor that helps cells divide, KIF11 turned out to be a major hub turned down whenever the Norrin beta catenin signal was weakened.
From faulty gene to dying vessel cells
The researchers next tested what happens when KIF11 is lost. In human retinal vessel cells grown in dishes, reducing KIF11 slowed their growth and led to cell death. Under the electron microscope, these cells showed telltale signs of increased self clean up structures and damaged, shrunken mitochondria, a pattern linked to a type of iron driven cell death called ferroptosis. Large scale measurements of RNA and proteins confirmed that protective systems that normally detoxify reactive oxygen molecules and prevent fat in cell membranes from going rancid were weakened, while markers of oxidative stress, iron buildup, and lipid peroxidation rose. Treating the cells with a small molecule that blocks ferroptosis restored many of these defects, suggesting that this death program is a central problem when KIF11 is missing.
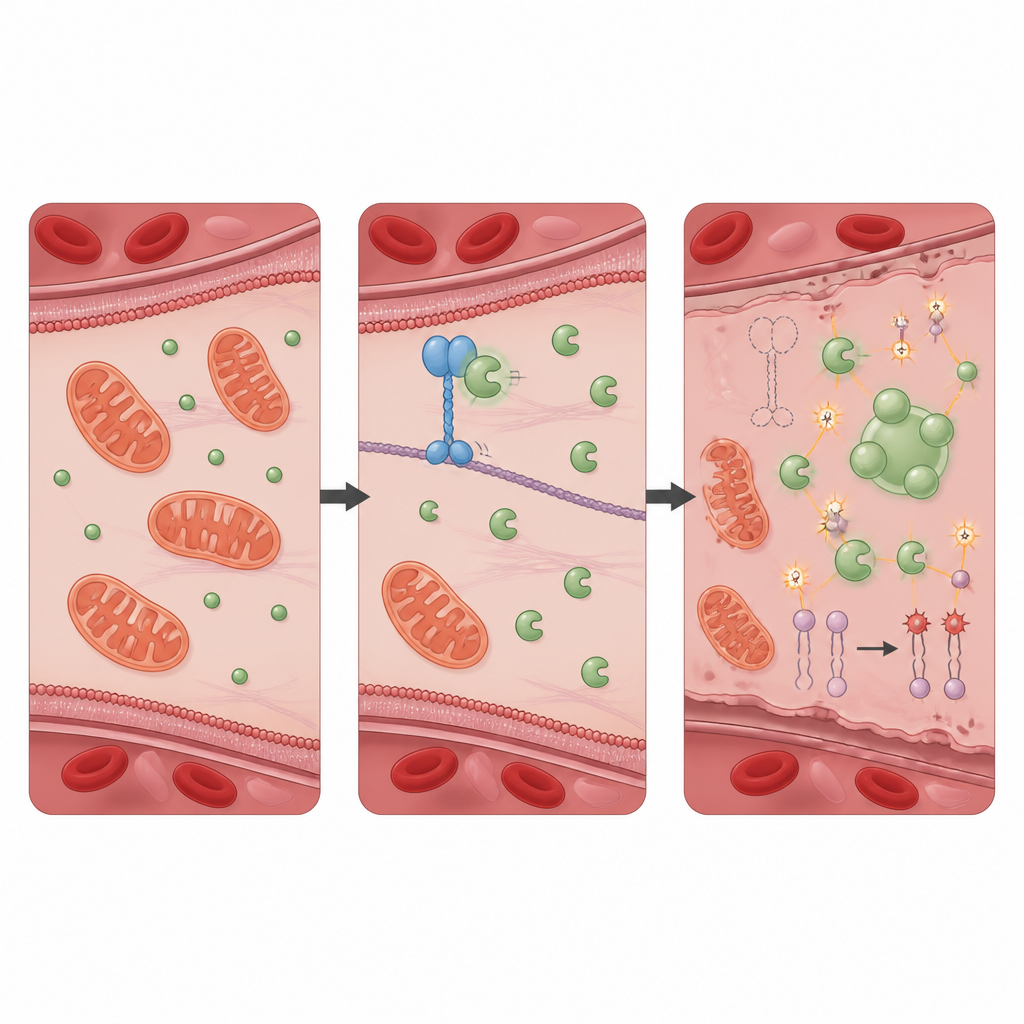
A protective partnership that keeps oxidative damage in check
To find out how KIF11 exerts this protective effect, the team searched for proteins it physically interacts with and identified PRDX1, a well known antioxidant enzyme that neutralizes harmful peroxides. In healthy cells, KIF11 binds PRDX1 and limits access of another protein, Src, which adds a phosphate group to PRDX1 at a specific site. When KIF11 is absent or truncated by patient mutations, Src can more easily modify PRDX1, prompting the enzyme molecules to join into larger clusters and form liquid like droplets inside the cell. This phase separation reduces PRDX1’s normal detoxifying activity, allowing oxidative stress to rise. Cells lacking PRDX1 alone showed a nearly identical pattern of self digestion, iron overload, membrane damage, and ferroptotic death as cells lacking KIF11, and these changes were also reversed by the ferroptosis blocker.
From molecular failure to faulty blood vessels in animals
Mouse models helped connect these microscopic events to real blood vessel defects. When the beta catenin signal was switched off only in endothelial cells, or when Kif11 itself was deleted in these cells, the growing retinal vessels in newborn mice stalled and were sparse, closely mimicking the human disease. The same mice showed more phosphorylated PRDX1, more signs of cellular self digestion, and shifts in ferroptosis related proteins in tissues rich in blood vessels. Giving young mice a ferroptosis inhibitor during the brief window of retinal vessel growth partially restored vessel coverage. Likewise, delivering extra KIF11, or a version of PRDX1 that cannot be phosphorylated at the critical site and so resists harmful clustering, improved vessel growth without obvious side effects.
What this means for protecting sight in children
Together, the findings outline a clear story: a signaling pathway that controls retinal vessel growth boosts KIF11, which in turn keeps the antioxidant PRDX1 active and evenly distributed, preventing runaway oxidative damage and iron driven cell death in vessel lining cells. When this pathway is weakened by inherited mutations, KIF11 levels fall, PRDX1 becomes over modified and clumps into droplets, and ferroptosis quietly erodes the cells that should build and maintain the retinal vasculature. While more work is needed before treatments reach patients, the study highlights two broad strategies that may help protect vision in this condition targeting ferroptosis itself, or stabilizing antioxidant defenses such as PRDX1 in the fragile developing eye.
Citation: Yang, M., Zhao, R., Peng, L. et al. KIF11 prevents retinal endothelial ferroptosis in familial exudative vitreoretinopathy by inhibiting phosphorylation-driven PRDX1 phase separation. Nat Commun 17, 4360 (2026). https://doi.org/10.1038/s41467-026-71009-7
Keywords: retinal blood vessels, ferroptosis, oxidative stress, familial exudative vitreoretinopathy, antioxidant enzymes