Clear Sky Science · sv
Optimera kvantkemiska simuleringar med ett hybridt kvantisering schema
Varför detta spelar roll för framtidens kemi
Design av nya läkemedel, batterier och material förlitar sig i allt högre grad på datorbaserade simuleringar av hur elektroner rör sig och växelverkar. Klassiska datorer har svårt med denna uppgift eftersom beräkningarna växer explosionsartat i kostnad när systemen blir större. Kvantdatorer lovar ett genombrott, men dagens kvantkemiska algoritmer använder olika interna språksystem för att beskriva elektroner, vilket försvårar att kombinera de bästa idéerna i ett enda arbetsflöde. I denna artikel presenteras ett sätt att smidigt översätta mellan dessa språk inom en och samma kvantkrets, vilket öppnar för effektivare simuleringar för en rad kemiska och materialvetenskapliga problem.
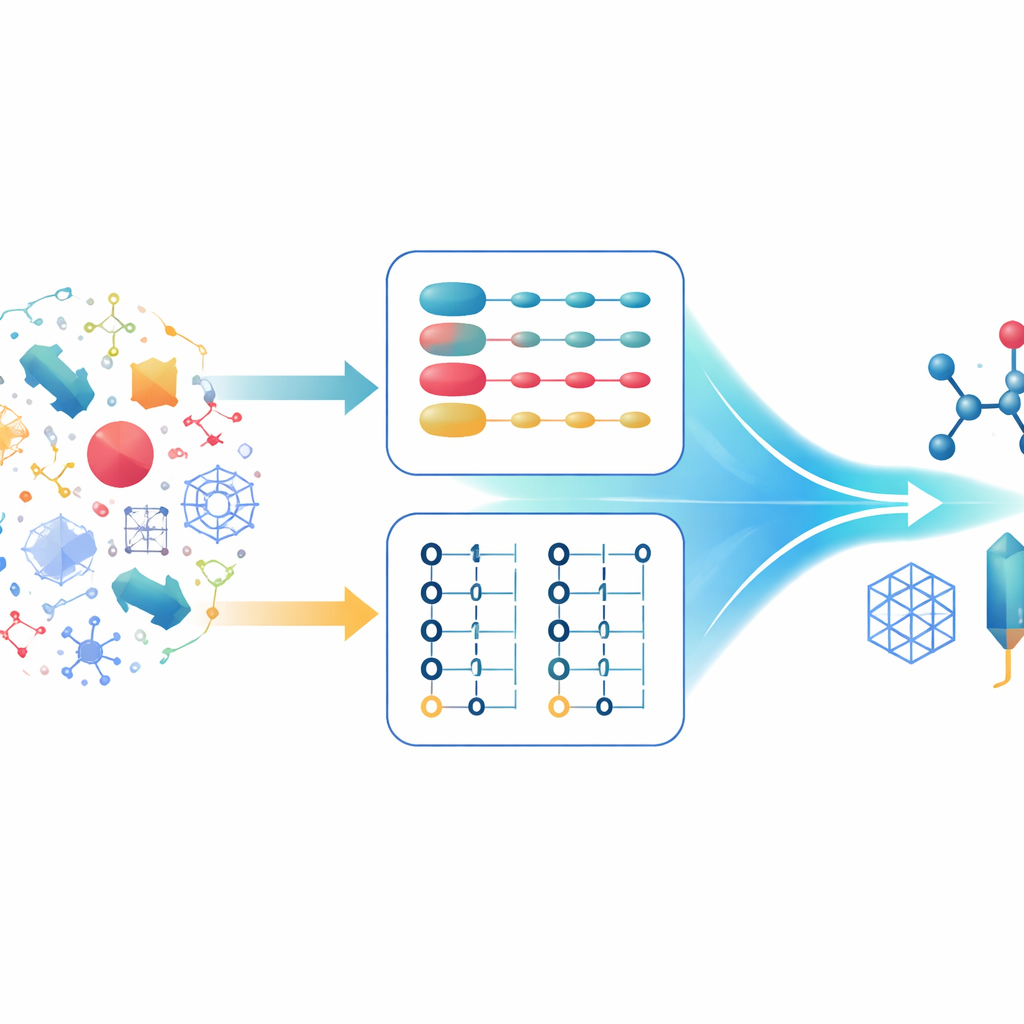
Två sätt att beskriva samma elektroner
Kvantkemister beskriver vanligtvis many-elektronsystem med två huvudsakliga formalismer. I den första spåras varje elektron individuellt och det övergripande tillståndet byggs upp från kombinationer av enelektrontillstånd. Detta är särskilt kraftfullt vid arbete med mycket regelbundna matematiska beskrivningar av rummet, såsom planvågor, vilka passar bra för utsträckta fasta ämnen. I den andra formalismen skiftar fokus från elektroner till om orbitaler är besatta eller tomma. Denna beskrivning respekterar naturligt regeln att två elektroner inte kan befinna sig i samma tillstånd och hanterar enkelt situationer där antalet elektroner ändras. Varje angreppssätt har sina styrkor och svagheter, och moderna kvantalgoritmer har optimerats noggrant kring det ena eller det andra, men inte båda samtidigt.
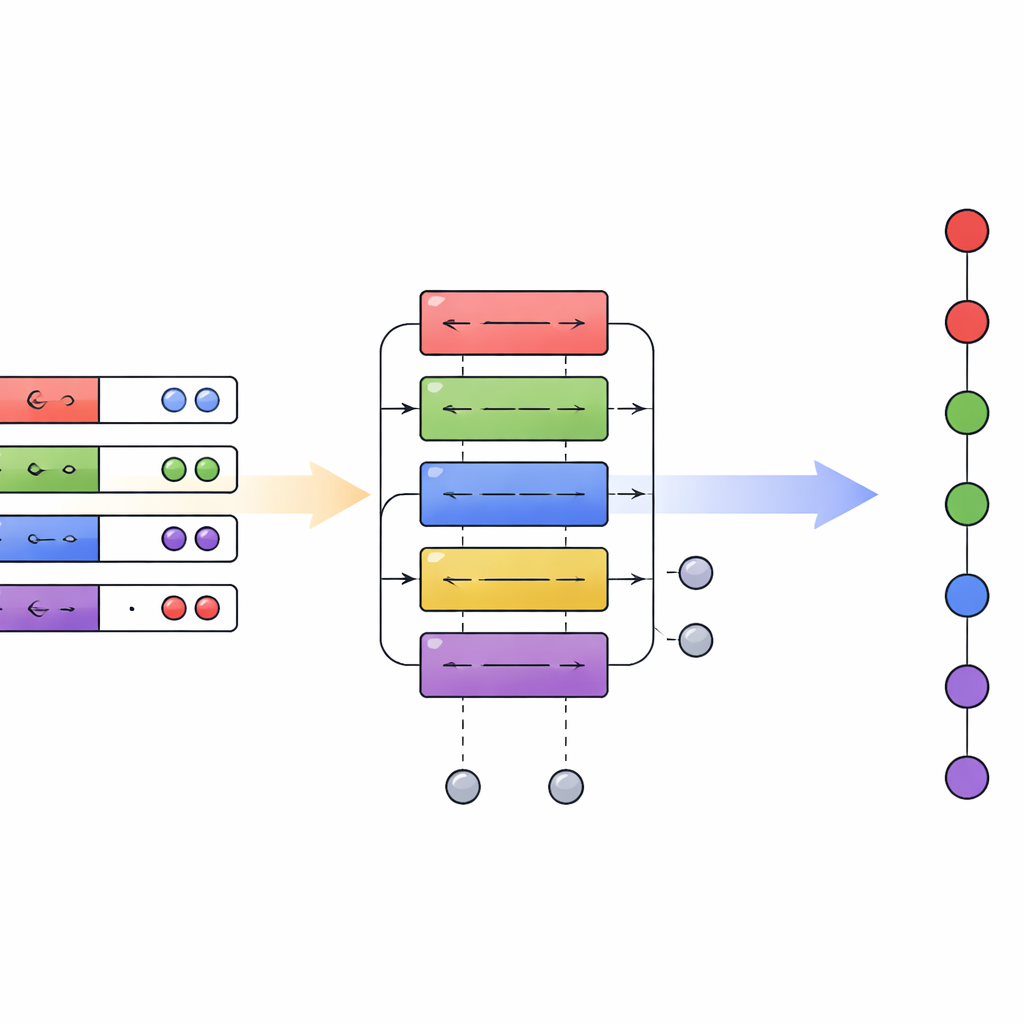
En kvant-"översättare" mellan kodningar
Författarna föreslår ett hybridt kvantisering schema som fungerar som en översättare mellan de två sätten att koda elektroner på en kvantdator. De bygger vidare på kompakta datalayouts som lagrar orbitalindex i binär form och visar att dessa delade strukturer möjliggör konversion mellan första kvantiseringen och en effektiv andra-quantiserad kodning med endast ett måttligt antal kvantlogiska grindar. Det centrala teoretiska resultatet, sats 1, bevisar att denna översättning kan utföras med ett antal grindar som växer endast något snabbare än linjärt med antalet elektroner och endast logaritmiskt med antalet orbitaler. Viktigt är att overheaden för att byta representation är liten jämfört med de vinster som uppnås genom att välja den bästa beskrivningen för varje del av en simulering.
Blanda och matcha för verkliga kemiska arbetsflöden
Med denna översättare visar artikeln hur man kan omkonstruera hela kvantsimuleringsarbetsflöden. För grundtillståndsberäkningar av molekyler och bulkmaterial kan man förbereda det elektroniska grundtillståndet i den andra-quantiserade form som är mest effektiv för molekylorbitaler, och sedan konvertera till första kvantiseringen för att mäta samlingar av elektroniska egenskaper med en teknik kallad klassiska skuggor (classical shadows). Denna strategi minskar kraftigt antalet gånger det dyra grundtillståndet behöver förberedas—med faktorer på tiotals till tusentals i författarnas numeriska tester för vanliga molekyler och stora basuppsättningar. För lokaliserade defekter eller adsorberade molekyler på ytor stöder metoden kombinationer av beskrivningar anpassade till det utsträckta fastämnet med mer kompakta orbitaler i regionen av intresse, vilket förbättrar uppskattningen av lokala observabler.
Bättre simuleringar av rörelse och ljus–materie-interaktion
Det hybrida schemat förbättrar också simuleringar där atomer rör sig eller elektroner läggs till och tas bort. I Born–Oppenheimer-molekylärdynamik behandlas elektroner kvantmekaniskt medan kärnorna rör sig enligt klassiska krafter härledda från det elektroniska tillståndet. Här möjliggör översättning till första-kvantiserad kodning effektivare kraftberäkningar från reducerade densitetsmatriser, vilket leder till stora besparingar i de upprepade mätningar som krävs vid varje tidssteg. För spektroskopiska och elektronjoniseringsproblem—där elektroner kan hoppa in eller ut ur ett material—är den underliggande tidsutvecklingen mest effektiv i den första-kvantiserade planvågsbeskrivningen, men de operationer som förändrar antalet elektroner passar naturligt i den andra-quantiserade vyn. Författarna visar hur man väver fram och tillbaka mellan dessa kodningar så att varje steg i beräkningen av Greens funktioner eller joniseringssannolikheter använder det mest ekonomiska verktyget.
En ny plan för kvantkemi på kvantdatorer
Sammantaget visar artikeln att ett omsorgsfullt utformat översättningslager mellan olika kvantkodningar kan ge breda, polynomskaliga effektivitetsvinster utan att kräva radikalt ny hårdvara. Genom att göra det praktiskt att kombinera första- och andra-quantiserade algoritmer inom en och samma krets lägger det hybrida kvantisering ramverket fram en mer flexibel plan för kvantkemi. När kvantprocessorer mognar kan denna förmåga att välja rätt representation i varje steg—istället för att vara låst till en—kraftigt minska resurserna som krävs för att simulera realistiska kemiska system och föra tillämpningar som noggrann reaktionsmodellering, materialupptäckt och avancerad spektroskopi närmare praktisk kvantfördel.
Citering: Ku, C., Chen, YC., Hu, A. et al. Optimizing quantum chemistry simulations with a hybrid quantization scheme. Commun Phys 9, 148 (2026). https://doi.org/10.1038/s42005-026-02577-9
Nyckelord: kvantkemi, kvantalgoritmer, elektronstruktur, materialsimulering, spektroskopi