Clear Sky Science · pl
Optymalizacja symulacji chemii kwantowej za pomocą hybrydowego schematu kwantyzacji
Dlaczego to ma znaczenie dla przyszłej chemii
Projektowanie nowych leków, baterii i materiałów coraz częściej opiera się na symulacjach komputerowych ruchu i oddziaływań elektronów. Komputery klasyczne mają z tym trudności, ponieważ koszty obliczeń gwałtownie rosną wraz ze wzrostem rozmiaru układów. Komputery kwantowe obiecują przełom, jednak współczesne algorytmy chemii kwantowej używają różnych „języków” wewnętrznych do opisu elektronów, co utrudnia połączenie najlepszych pomysłów w jednym przepływie pracy. Artykuł przedstawia sposób na płynne tłumaczenie między tymi językami wewnątrz jednego obwodu kwantowego, umożliwiając wydajniejsze symulacje dla szerokiego zakresu problemów chemicznych i materiałowych.
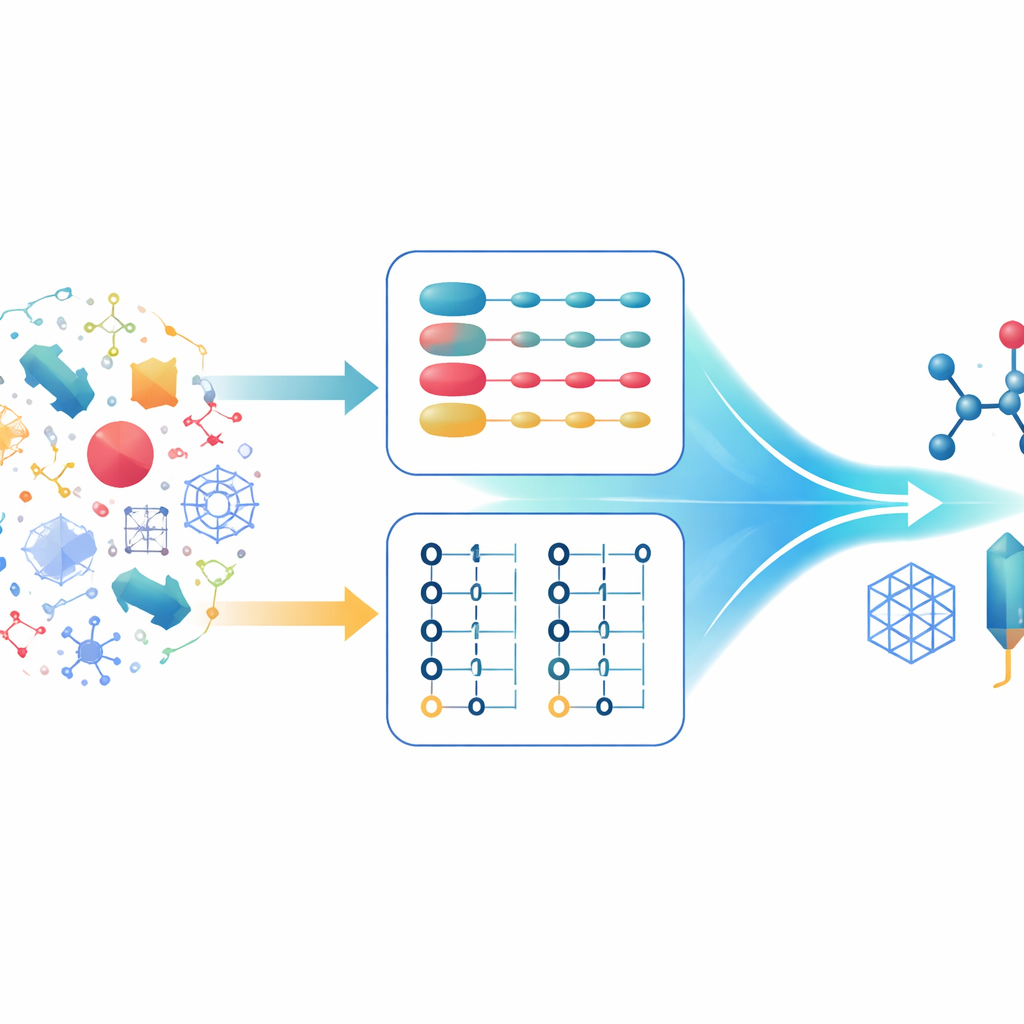
Dwa sposoby opisu tych samych elektronów
Chemicy kwantowi zazwyczaj opisują układy wieloelektronowe, stosując dwie główne formalizmy. W pierwszym każdy elektron jest śledzony indywidualnie, a stan całkowity budowany jest z kombinacji stanów pojedynczych elektronów. Jest to szczególnie efektywne przy pracy z regularnymi matematycznymi opisami przestrzeni, takimi jak fale płaskie, korzystnymi dla układów rozciągłych. W drugim formalizmie uwaga przesuwa się z elektronów na obsadzone lub puste orbitale. Ten opis naturalnie respektuje reguły wykluczania oraz łatwo radzi sobie z sytuacjami, gdy liczba elektronów się zmienia. Każde podejście ma swoje mocne i słabe strony, a nowoczesne algorytmy kwantowe są zwykle optymalizowane wokół jednego z nich, a nie wokół obu jednocześnie.
Kwantowy „tłumacz” między kodowaniami
Autorzy proponują hybrydowy schemat kwantyzacji działający jak tłumacz między dwoma sposobami kodowania elektronów na komputerze kwantowym. Bazują na kompaktowych układach danych, które przechowują indeksy orbitali w postaci binarnej, i pokazują, że te wspólne struktury pozwalają na konwersję między opisem pierwszego kwantowania a efektywnym kodowaniem drugiego kwantowania przy użyciu jedynie umiarkowanej liczby bramek kwantowych. Kluczowy wynik teoretyczny, Twierdzenie 1, dowodzi, że to tłumaczenie można wykonać z liczbą bramek rosnącą jedynie nieznacznie szybciej niż liniowo względem liczby elektronów i jedynie logarytmicznie względem liczby orbitali. Co ważne, narzut związany ze zmianą reprezentacji jest niewielki w porównaniu z oszczędnościami uzyskanymi przez wybór najlepszej reprezentacji dla poszczególnych części symulacji.
Mieszanie i dopasowywanie w rzeczywistych przepływach pracy
Posiadając takiego tłumacza, artykuł pokazuje, jak przeprojektować pełne przepływy pracy symulacji kwantowych. Przy obliczeniach stanu podstawowego cząsteczek i materiałów stałych można przygotować stan podstawowy elektronów w formie drugiego kwantowania, najbardziej efektywnej dla orbitali molekularnych, a następnie przełączyć na pierwsze kwantowanie, aby zmierzyć zestawy wielkości elektronowych za pomocą techniki zwanej klasycznymi cieniami. Strategia ta znacząco redukuje liczbę przygotowań kosztownego stanu podstawowego — w testach numerycznych autorów liczba ta zmniejszyła się od kilkudziesięciu do kilku tysięcy razy dla typowych cząsteczek i dużych baz. Dla lokalizowanych defektów lub cząsteczek adsorbowanych na powierzchniach metoda umożliwia łączenie opisów dopasowanych do rozciągłego kryształu z bardziej zwartymi orbitalami w pobliżu obszaru zainteresowania, poprawiając estymację obserwowalnych lokalnych.
Lepsze symulacje ruchu i oddziaływania światła z materią
Hybrydowy schemat poprawia także symulacje, w których atomy się przemieszczają lub elektrony są dodawane i usuwane. W klasycznej metodzie Borna–Oppenheimera elektrony traktowane są kwantowo, podczas gdy jądra poruszają się według klasycznych sił wyprowadzonych ze stanu elektronowego. Tutaj przejście do kodowania pierwszego kwantowania umożliwia bardziej efektywne obliczanie sił z macierzy gęstości zredukowanej, prowadząc do dużych oszczędności w powtarzanych pomiarach wymaganych na każdym kroku czasowym. W problemach spektroskopowych i jonizacji elektronowej — gdzie elektrony mogą wchodzić lub wychodzić z materiału — ewolucja czasowa jest najwydajniejsza w opisie pierwszego kwantowania w falach płaskich, natomiast operacje zmieniające liczbę elektronów naturalnie pasują do widoku drugiego kwantowania. Autorzy pokazują, jak przemykać między tymi kodowaniami, tak aby każdy etap obliczania funkcji Greena czy prawdopodobieństw jonizacji używał najbardziej ekonomicznego narzędzia.
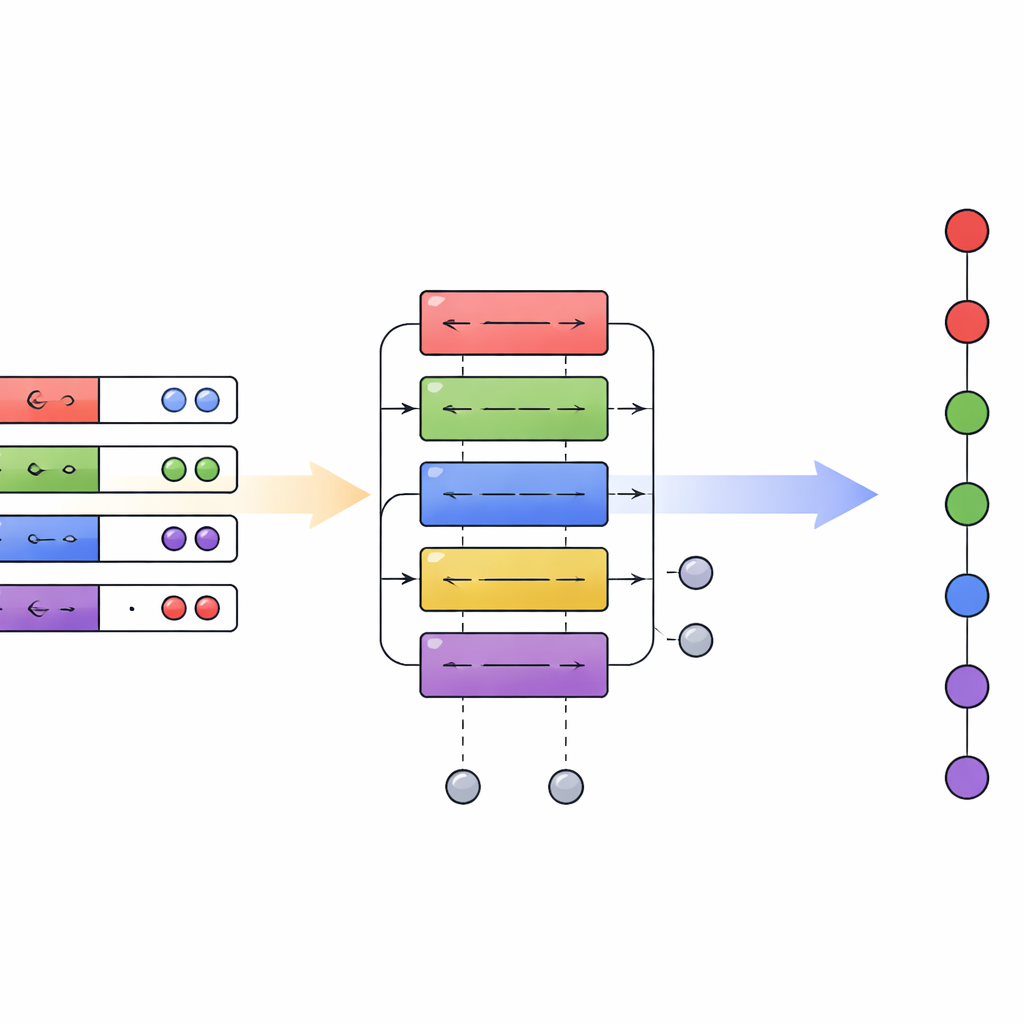
Nowy plan dla chemii kwantowej na komputerach kwantowych
Podsumowując, artykuł wykazuje, że starannie zaprojektowana warstwa tłumacząca między różnymi kodowaniami kwantowymi może przynieść szerokie, wielomianowe zyski wydajności bez konieczności radykalnej zmiany sprzętu. Umożliwiając praktyczne łączenie algorytmów pierwszego i drugiego kwantowania w jednym obwodzie, ramy hybrydowej kwantyzacji przedstawiają elastyczniejszy plan dla chemii kwantowej. W miarę dojrzewania procesorów kwantowych zdolność wyboru odpowiedniej reprezentacji na każdym etapie — zamiast bycia ograniczonym do jednej — może znacznie zmniejszyć zasoby potrzebne do symulacji realistycznych układów chemicznych, przybliżając zastosowania takie jak dokładne modelowanie reakcji, odkrywanie materiałów i zaawansowana spektroskopia do praktycznej przewagi kwantowej.
Cytowanie: Ku, C., Chen, YC., Hu, A. et al. Optimizing quantum chemistry simulations with a hybrid quantization scheme. Commun Phys 9, 148 (2026). https://doi.org/10.1038/s42005-026-02577-9
Słowa kluczowe: chemia kwantowa, algorytmy kwantowe, struktura elektronów, symulacja materiałów, spektroskopia