Clear Sky Science · es
Optimización de simulaciones de química cuántica con un esquema de cuantización híbrido
Por qué esto importa para la química del futuro
El diseño de nuevos medicamentos, baterías y materiales depende cada vez más de simulaciones por ordenador de cómo se mueven e interactúan los electrones. Los ordenadores clásicos tienen dificultades con esta tarea porque los cálculos crecen exponencialmente en coste conforme aumentan los sistemas. Los ordenadores cuánticos prometen un avance, pero los algoritmos de química cuántica actuales usan distintos lenguajes internos para describir a los electrones, lo que dificulta combinar las mejores ideas en un único flujo de trabajo. Este artículo presenta una forma de traducir suavemente entre esos lenguajes dentro de un mismo circuito cuántico, desbloqueando simulaciones más eficientes para una amplia gama de problemas químicos y de materiales.
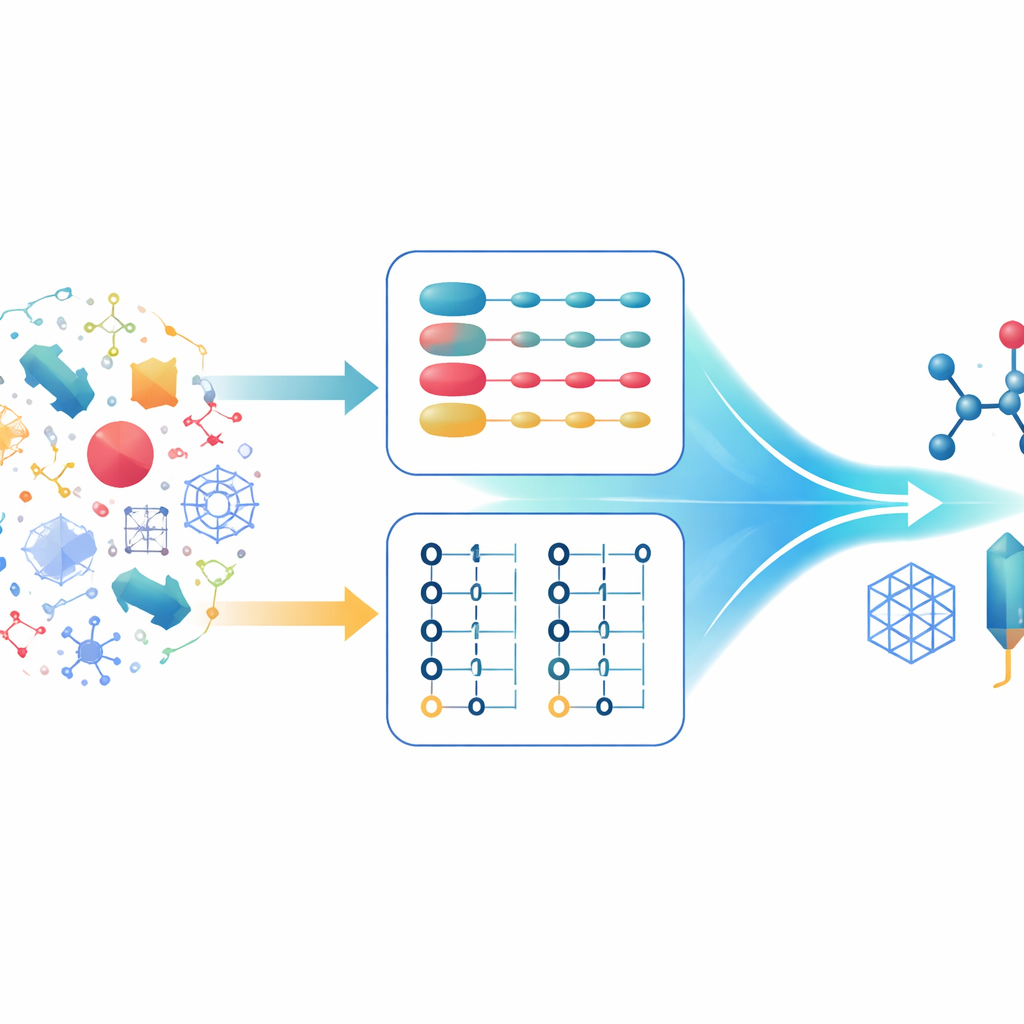
Dos formas de describir los mismos electrones
Los químicos cuánticos suelen describir sistemas de muchos electrones con dos formalismos principales. En el primero, cada electrón se sigue individualmente y el estado global se construye a partir de combinaciones de estados de un único electrón. Esto es especialmente potente al trabajar con descripciones matemáticas muy regulares del espacio, como las ondas planas, que son adecuadas para sólidos extendidos. En el segundo formalismo, el foco cambia desde los electrones a los orbitales ocupados o vacíos. Esta descripción respeta de forma natural la regla de que no pueden ocupar el mismo estado dos electrones y maneja fácilmente situaciones donde cambia el número de electrones. Cada enfoque tiene fortalezas y debilidades, y los algoritmos cuánticos modernos se han optimizado cuidadosamente en torno a uno u otro, pero no a ambos a la vez.
Un “traductor” cuántico entre codificaciones
Los autores proponen un esquema de cuantización híbrido que actúa como traductor entre las dos formas de codificar electrones en un ordenador cuántico. Se basan en distribuciones de datos compactas que almacenan índices de orbitales en forma binaria y muestran que estas estructuras compartidas permiten la conversión entre la primera cuantización y una codificación eficiente de segunda cuantización usando solo un número moderado de puertas lógicas cuánticas. El resultado teórico clave, el Teorema 1, demuestra que esta traducción puede realizarse con un número de puertas que crece solo ligeramente más rápido que linealmente con el número de electrones y solo logarítmicamente con el número de orbitales. Es importante que la sobrecarga de cambiar de representación sea pequeña en comparación con los ahorros obtenidos al elegir la mejor descripción para cada parte de una simulación.
Combinar y adaptar para flujos de trabajo químicos reales
Con este traductor, el artículo muestra cómo reingeniarizar flujos completos de simulación cuántica. Para cálculos del estado fundamental de moléculas y materiales, se puede preparar el estado electrónico fundamental en la forma de segunda cuantización que resulta más eficiente para orbitales moleculares y luego convertir a la primera cuantización para medir conjuntos de propiedades electrónicas usando una técnica llamada sombras clásicas. Esta estrategia reduce drásticamente el número de veces que hay que preparar el costoso estado fundamental: en las pruebas numéricas de los autores, las reducciones van de decenas a miles para moléculas comunes y grandes conjuntos de base. Para defectos localizados o moléculas adsorbidas en superficies, el método permite combinar descripciones adaptadas al sólido extendido con orbitales más compactos cerca de la región de interés, mejorando la estimación de observables locales.
Mejores simulaciones de movimiento y de interacción luz‑materia
El esquema híbrido también mejora las simulaciones en las que los átomos se mueven o se añaden/quitan electrones. En la dinámica molecular Born–Oppenheimer, los electrones se tratan cuánticamente mientras los núcleos se mueven según fuerzas clásicas derivadas del estado electrónico. Aquí, traducir a la codificación de primera cuantización permite cálculos de fuerzas más eficientes a partir de matrices de densidad reducidas, lo que conlleva grandes ahorros en las mediciones repetidas requeridas en cada paso temporal. Para problemas de espectroscopía e ionización electrónica —donde los electrones pueden entrar o salir de un material— la evolución temporal subyacente es más eficiente en la descripción de ondas planas de primera cuantización, pero las operaciones que cambian el número de electrones encajan naturalmente en la visión de segunda cuantización. Los autores muestran cómo entrelazar ida y vuelta entre estas codificaciones para que cada paso del cálculo de funciones de Green o probabilidades de ionización use la herramienta más económica.
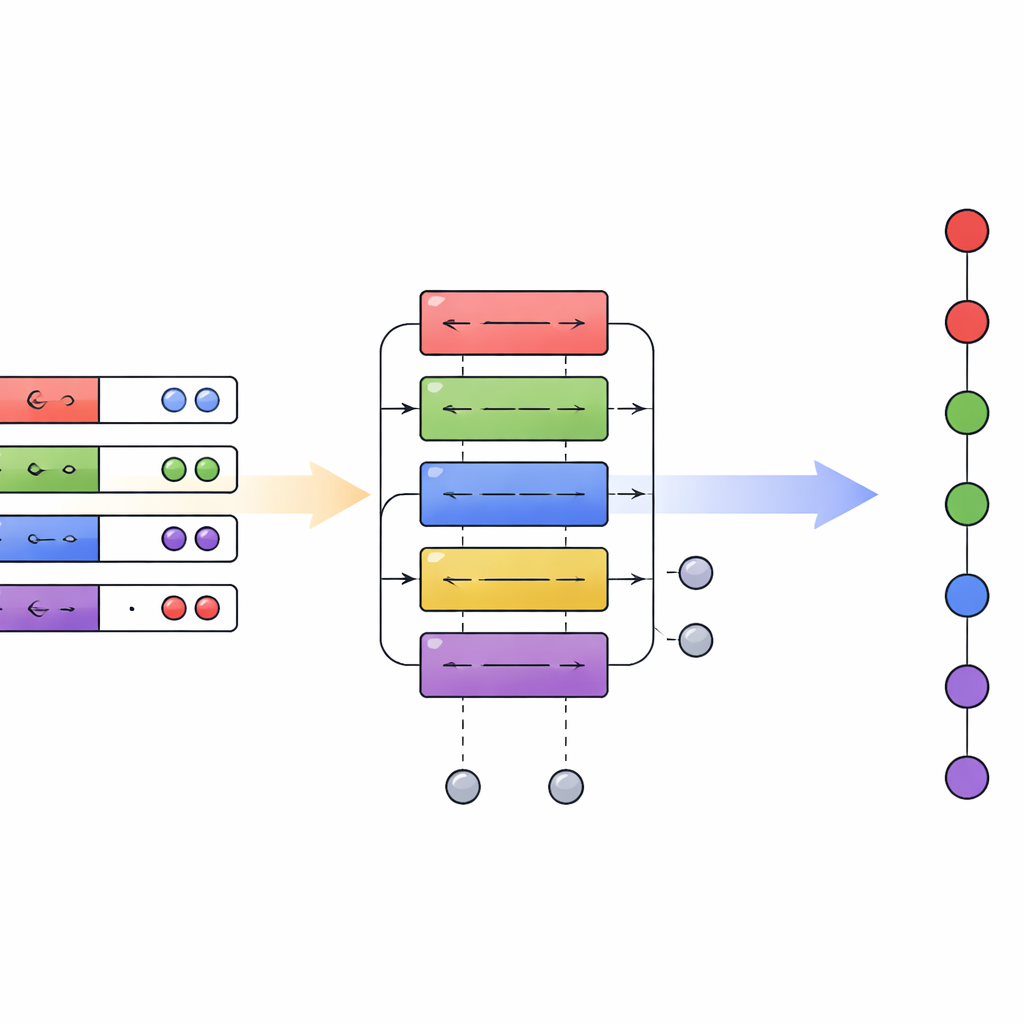
Un nuevo plan para la química cuántica en ordenadores cuánticos
En conjunto, el artículo demuestra que una capa de traducción diseñada con cuidado entre diferentes codificaciones cuánticas puede producir ganancias de eficiencia a escala polinómica sin requerir hardware radicalmente nuevo. Al hacer práctica la combinación de algoritmos de primera y segunda cuantización dentro de un único circuito, el marco de cuantización híbrido traza un plan más flexible para la química cuántica. A medida que los procesadores cuánticos maduren, esta capacidad de elegir la representación adecuada en cada etapa —en lugar de quedarse bloqueado en una sola— podría reducir sustancialmente los recursos necesarios para simular sistemas químicos realistas, acercando aplicaciones como el modelado preciso de reacciones, el descubrimiento de materiales y la espectroscopía avanzada a una ventaja cuántica práctica.
Cita: Ku, C., Chen, YC., Hu, A. et al. Optimizing quantum chemistry simulations with a hybrid quantization scheme. Commun Phys 9, 148 (2026). https://doi.org/10.1038/s42005-026-02577-9
Palabras clave: química cuántica, algoritmos cuánticos, estructura electrónica, simulación de materiales, espectroscopía