Clear Sky Science · pt
Otimização de simulações de química quântica com um esquema híbrido de quantização
Por que isso importa para a química do futuro
Projetar novos medicamentos, baterias e materiais depende cada vez mais de simulações computacionais de como os elétrons se movem e interagem. Computadores clássicos enfrentam dificuldades nessa tarefa porque os cálculos crescem exponencialmente em custo à medida que os sistemas aumentam. Computadores quânticos prometem um avanço, mas os algoritmos de química quântica atuais usam linguagens internas diferentes para descrever elétrons, o que dificulta combinar as melhores ideias em um único fluxo de trabalho. Este artigo introduz uma forma de traduzir suavemente entre essas linguagens dentro de um único circuito quântico, liberando simulações mais eficientes para uma ampla gama de problemas químicos e de materiais.
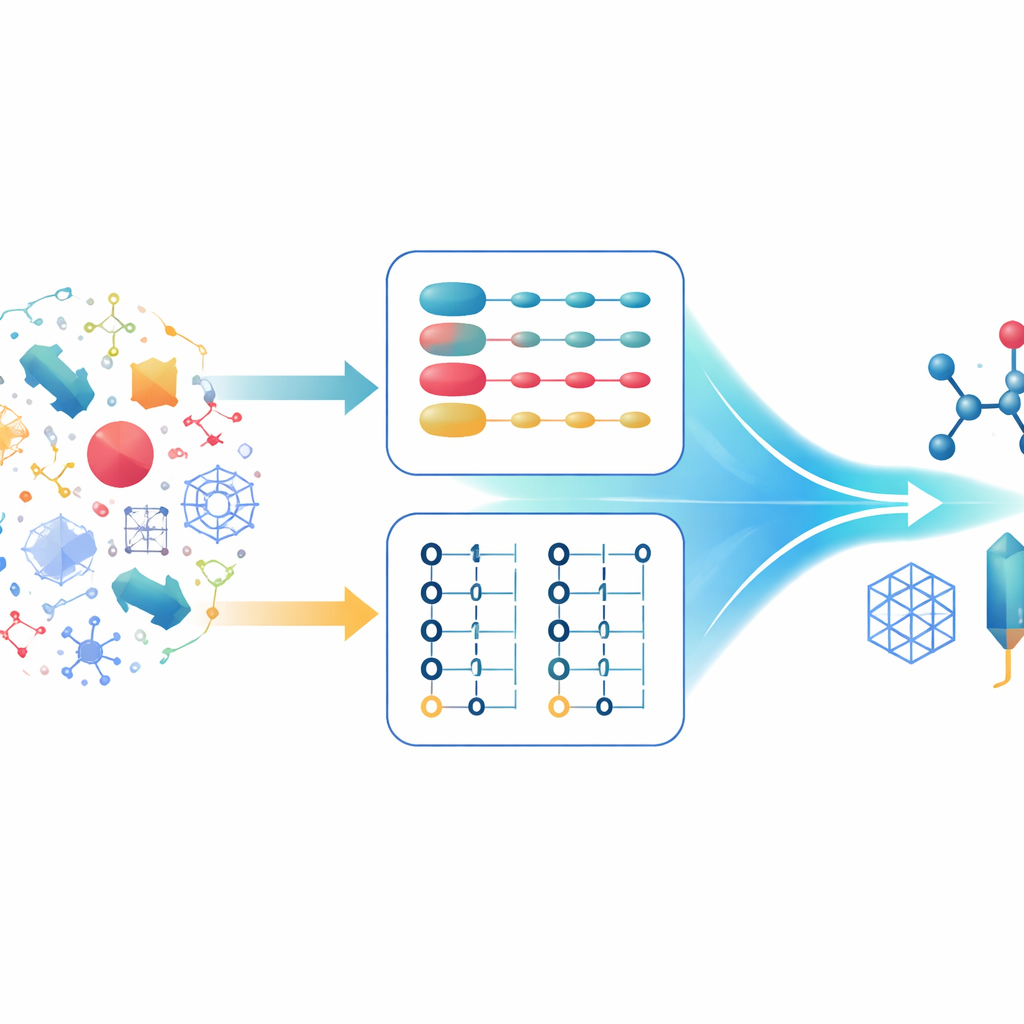
Duas maneiras de descrever os mesmos elétrons
Químicos quânticos tipicamente descrevem sistemas de muitos elétrons usando dois formalismos principais. No primeiro, cada elétron é acompanhado individualmente, e o estado global é construído a partir de combinações de estados de um único elétron. Isso é especialmente poderoso ao trabalhar com descrições matemáticas altamente regulares do espaço, como ondas planas, que se adaptam bem a sólidos estendidos. No segundo formalismo, o foco muda dos elétrons para orbitais ocupados ou vazios. Essa descrição respeita naturalmente as regras de que dois elétrons não podem ocupar o mesmo estado e lida facilmente com situações em que o número de elétrons varia. Cada abordagem tem pontos fortes e fracos, e algoritmos quânticos modernos foram cuidadosamente otimizados em torno de um ou outro, mas não de ambos simultaneamente.
Um “tradutor” quântico entre codificações
Os autores propõem um esquema de quantização híbrido que funciona como um tradutor entre as duas maneiras de codificar elétrons em um computador quântico. Eles se baseiam em layouts de dados compactos que armazenam índices de orbitais em forma binária e mostram que essas estruturas compartilhadas permitem a conversão entre a primeira quantização e uma codificação eficiente em segunda quantização usando apenas um número modesto de portas lógicas quânticas. O resultado teórico chave, o Teorema 1, prova que essa tradução pode ser feita com uma contagem de portas que cresce apenas um pouco mais rápido que linearmente com o número de elétrons, e apenas logaritmicamente com o número de orbitais. Importante, o overhead de mudar de representação é pequeno em comparação com as economias obtidas ao escolher a melhor descrição para cada parte de uma simulação.
Misturar e combinar para fluxos de trabalho químicos reais
Armados com esse tradutor, os autores mostram como reengenheirar fluxos de trabalho completos de simulação quântica. Para cálculos do estado fundamental de moléculas e materiais em bloco, pode-se preparar o estado eletrônico fundamental na forma de segunda quantização que é mais eficiente para orbitais moleculares, e então converter para a primeira quantização para medir coleções de propriedades eletrônicas usando uma técnica chamada sombras clássicas (classical shadows). Essa estratégia reduz drasticamente o número de vezes que o estado fundamental caro precisa ser preparado — por fatores de dezenas a milhares nos testes numéricos dos autores para moléculas comuns e grandes bases de orbitais. Para defeitos localizados ou moléculas adsorvidas em superfícies, o método permite combinar descrições adaptadas ao sólido estendido com orbitais mais compactos próximos à região de interesse, melhorando a estimativa de observáveis locais.
Melhores simulações de movimento e luz–matéria
O esquema híbrido também melhora simulações em que átomos se movem ou elétrons são adicionados e removidos. Em dinâmica molecular de Born–Oppenheimer, os elétrons são tratados quanticamente enquanto os núcleos se movem segundo forças clássicas derivadas do estado eletrônico. Aqui, traduzir para a codificação de primeira quantização possibilita cálculos de forças mais eficientes a partir de matrizes de densidade reduzidas, levando a grandes economias nas medições repetidas exigidas a cada passo de tempo. Para problemas de espectroscopia e ionização eletrônica — onde elétrons podem entrar ou sair de um material — a evolução temporal subjacente é mais eficiente na descrição de ondas planas em primeira quantização, mas as operações que mudam o número de elétrons se encaixam naturalmente na visão de segunda quantização. Os autores mostram como alternar entre essas codificações de modo que cada etapa do cálculo de funções de Green ou probabilidades de ionização use a ferramenta mais econômica.
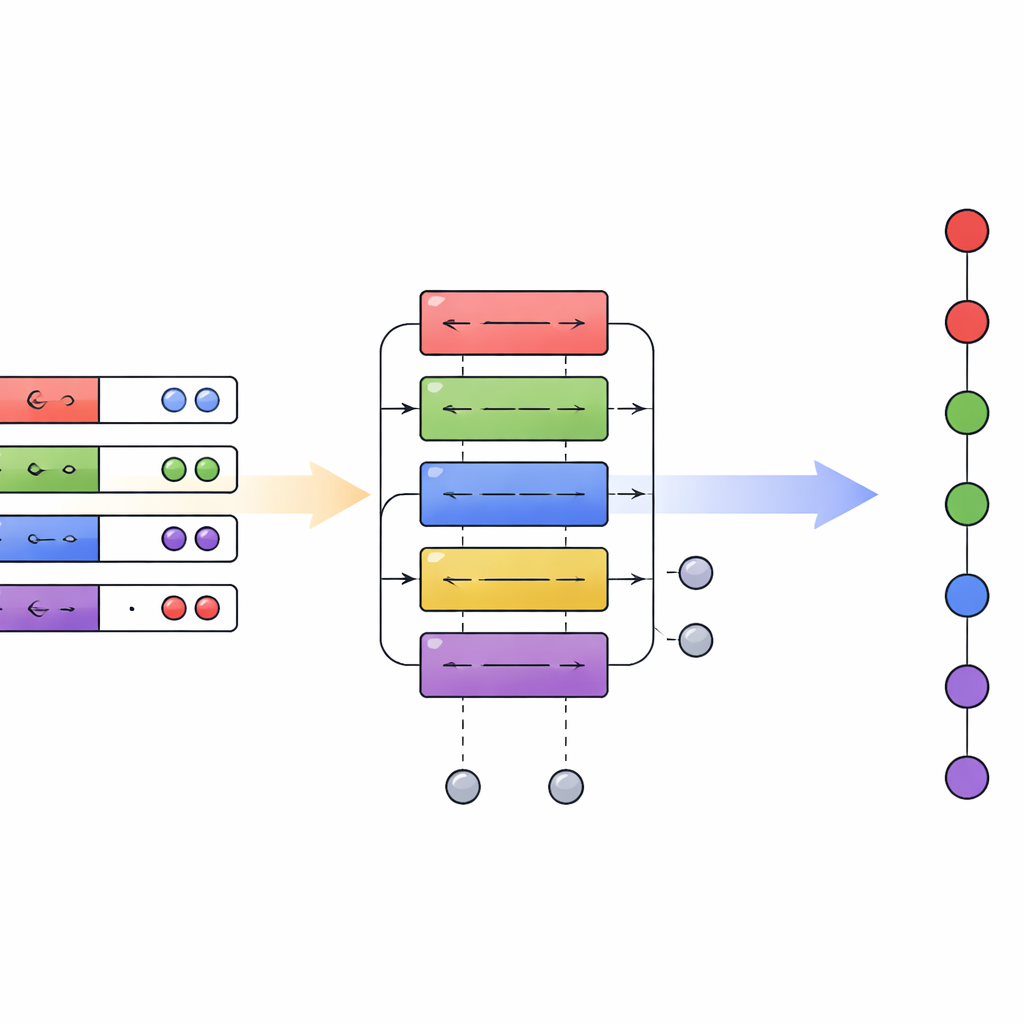
Um novo roteiro para química quântica em computadores quânticos
Em síntese, o artigo demonstra que uma camada de tradução cuidadosamente projetada entre diferentes codificações quânticas pode proporcionar ganhos de eficiência em escala polinomial sem exigir hardware radicalmente novo. Tornando prático combinar algoritmos de primeira e segunda quantização dentro de um único circuito, a estrutura de quantização híbrida traça um roteiro mais flexível para a química quântica. À medida que os processadores quânticos amadurecem, essa capacidade de escolher a representação certa em cada etapa — em vez de ficar preso a uma só — pode reduzir substancialmente os recursos necessários para simular sistemas químicos realistas, aproximando aplicações como modelagem precisa de reações, descoberta de materiais e espectroscopia avançada de uma vantagem quântica prática.
Citação: Ku, C., Chen, YC., Hu, A. et al. Optimizing quantum chemistry simulations with a hybrid quantization scheme. Commun Phys 9, 148 (2026). https://doi.org/10.1038/s42005-026-02577-9
Palavras-chave: química quântica, algoritmos quânticos, estrutura eletrônica, simulação de materiais, espectroscopia