Clear Sky Science · fr
Optimisation des simulations de chimie quantique avec un schéma de quantification hybride
Pourquoi cela compte pour la chimie de demain
La conception de nouveaux médicaments, batteries et matériaux dépend de plus en plus de simulations informatiques du mouvement et des interactions des électrons. Les ordinateurs classiques peinent pour cette tâche car le coût des calculs explose quand les systèmes grandissent. Les ordinateurs quantiques promettent une percée, mais les algorithmes de chimie quantique actuels utilisent des langages internes différents pour décrire les électrons, ce qui complique la combinaison des meilleures idées au sein d’un même flux de travail. Cet article présente une méthode pour traduire en douceur entre ces langages au sein d’un même circuit quantique, débloquant des simulations plus efficaces pour un large éventail de problèmes chimiques et matériaux.
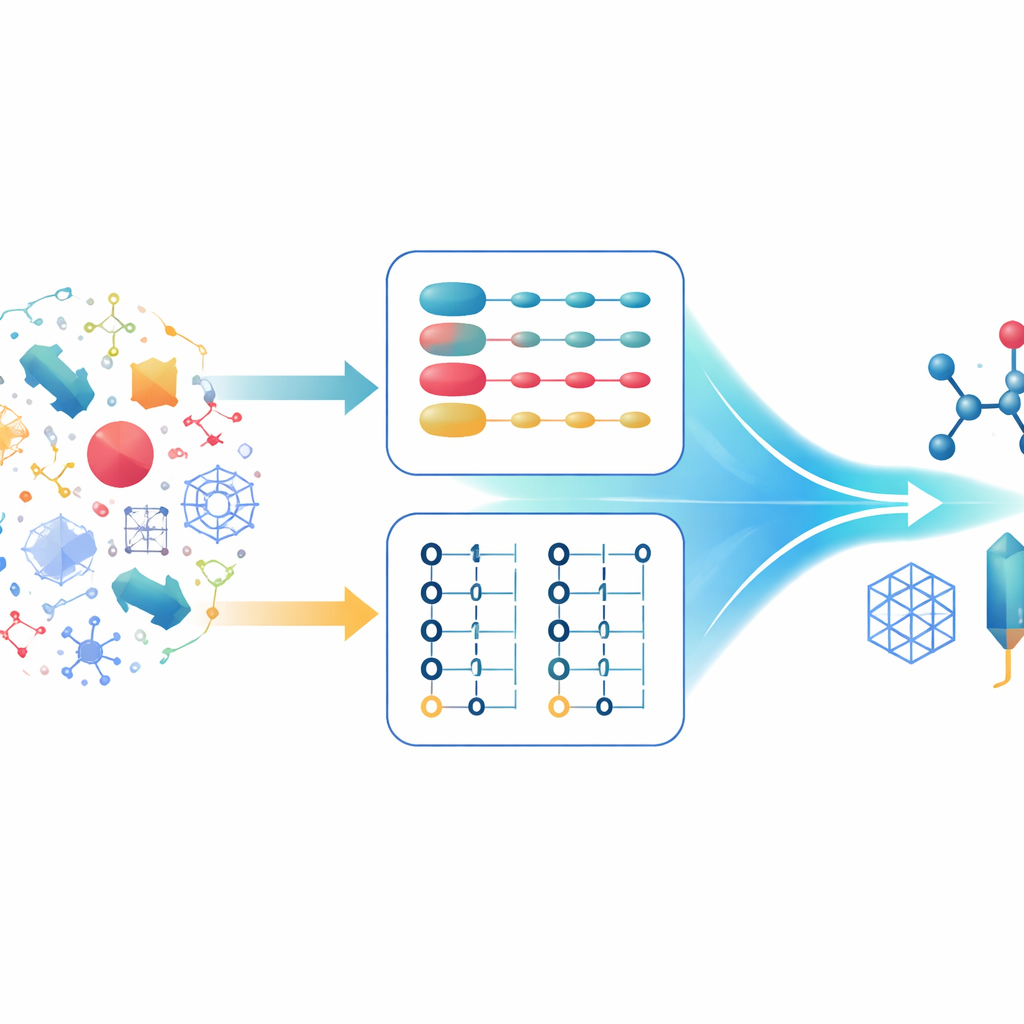
Deux manières de décrire les mêmes électrons
Les chimistes quantiques décrivent typiquement les systèmes à plusieurs électrons à l’aide de deux formalisms principaux. Dans le premier, chaque électron est suivi individuellement, et l’état global se construit à partir de combinaisons d’états monélectroniques. Ceci est particulièrement puissant lorsqu’on travaille avec des descriptions mathématiques très régulières de l’espace, comme les ondes planes, adaptées aux solides étendus. Dans le second formalisme, l’attention se porte sur les orbitales, occupées ou vides. Cette description respecte naturellement la règle selon laquelle deux électrons ne peuvent occuper le même état et gère facilement les situations où le nombre d’électrons varie. Chaque approche a ses forces et ses faiblesses, et les algorithmes quantiques modernes ont été optimisés autour de l’une ou l’autre, mais pas des deux simultanément.
Un « traducteur » quantique entre encodages
Les auteurs proposent un schéma de quantification hybride qui agit comme un traducteur entre les deux manières d’encoder les électrons sur un ordinateur quantique. Ils s’appuient sur des agencements compacts de données qui stockent les indices d’orbitales en binaire, et montrent que ces structures partagées permettent la conversion entre la première quantification et un encodage efficace en seconde quantification en n’utilisant qu’un nombre modéré de portes logiques quantiques. Le résultat théorique clé, le Théorème 1, démontre que cette traduction peut être réalisée avec un nombre de portes qui croît seulement légèrement plus vite que linéairement avec le nombre d’électrons, et seulement logarithmiquement avec le nombre d’orbitales. Il est important de noter que le surcoût lié au changement de représentation est faible par rapport aux économies obtenues en choisissant la meilleure description pour chaque partie d’une simulation.
Combiner les approches pour des flux de travail chimiques réels
Grâce à ce traducteur, l’article montre comment réingénieriser des flux de simulation quantique complets. Pour les calculs de l’état fondamental de molécules et de matériaux en vrac, on peut préparer l’état fondamental électronique sous la forme en seconde quantification la plus efficace pour les orbitales moléculaires, puis convertir en première quantification pour mesurer des collections de propriétés électroniques à l’aide d’une technique appelée ombres classiques (classical shadows). Cette stratégie réduit fortement le nombre de préparations coûteuses de l’état fondamental — de facteurs allant de dizaines à des milliers dans les tests numériques des auteurs pour des molécules courantes et de grands jeux de bases. Pour des défauts localisés ou des molécules adsorbées sur des surfaces, la méthode permet de combiner des descriptions adaptées au solide étendu avec des orbitales plus compactes près de la région d’intérêt, améliorant l’estimation des observables locaux.
Meilleures simulations de mouvement et d’interaction lumière-matière
Le schéma hybride améliore également les simulations où les atomes se déplacent ou des électrons sont ajoutés ou retirés. Dans la dynamique moléculaire Born–Oppenheimer, les électrons sont traités quantiquement tandis que les noyaux se déplacent sous l’action de forces classiques dérivées de l’état électronique. Ici, la traduction vers l’encodage en première quantification permet des calculs de forces plus efficaces à partir de matrices de densité réduites, entraînant d’importantes économies dans les mesures répétées requises à chaque pas de temps. Pour les problèmes de spectroscopie et d’ionisation électronique — où des électrons peuvent entrer ou sortir d’un matériau — l’évolution temporelle sous-jacente est la plus efficace dans la description en ondes planes de la première quantification, mais les opérations qui changent le nombre d’électrons s’insèrent naturellement dans la vision de la seconde quantification. Les auteurs montrent comment naviguer entre ces encodages afin que chaque étape du calcul des fonctions de Green ou des probabilités d’ionisation utilise l’outil le plus économique.
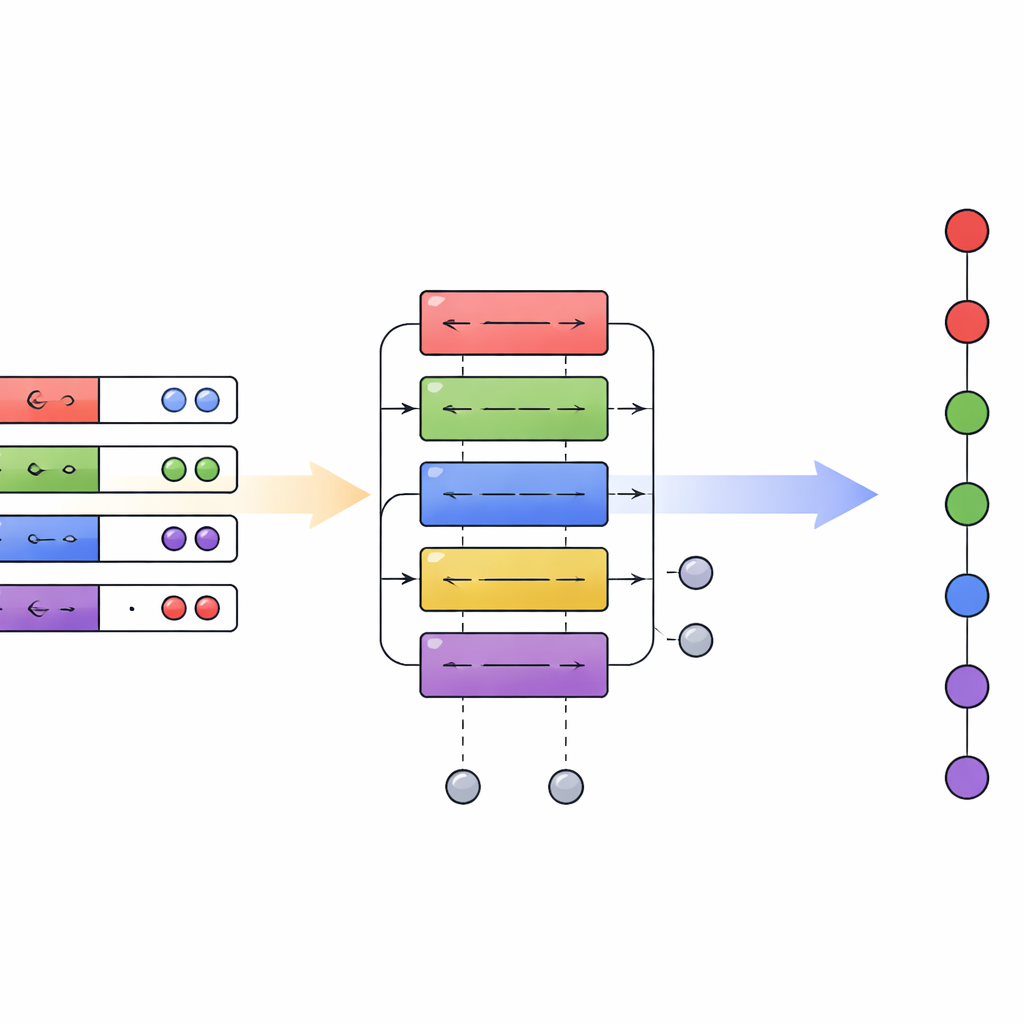
Un nouveau plan pour la chimie quantique sur ordinateurs quantiques
Au total, l’article démontre qu’une couche de traduction soigneusement conçue entre différents encodages quantiques peut offrir des gains d’efficacité à grande échelle polynomiale sans nécessiter un matériel radicalement nouveau. En rendant pratique la combinaison d’algorithmes en première et seconde quantification dans un même circuit, le cadre de quantification hybride propose un plan plus flexible pour la chimie quantique. À mesure que les processeurs quantiques mûriront, cette capacité à choisir la représentation adaptée à chaque étape — plutôt que d’être enfermé dans une seule — pourrait réduire sensiblement les ressources nécessaires pour simuler des systèmes chimiques réalistes, rapprochant des applications comme la modélisation précise de réactions, la découverte de matériaux et la spectroscopie avancée d’un avantage quantique pratique.
Citation: Ku, C., Chen, YC., Hu, A. et al. Optimizing quantum chemistry simulations with a hybrid quantization scheme. Commun Phys 9, 148 (2026). https://doi.org/10.1038/s42005-026-02577-9
Mots-clés: chimie quantique, algorithmes quantiques, structure électronique, simulation de matériaux, spectroscopie