Clear Sky Science · sv
Ny proteolytisk posttranslationell modifiering i spänningsstyrd kaliumkanal KCNQ2
Varför dessa små hjärnportar spelar roll
Våra tankar, rörelser och till och med nyfödda barns första skrik bygger på miljarder nervceller som avfyrar i noggrant tajmade mönster. Dessa elektriska signaler styrs av mikroskopiska ”portar” i cellmembranet som släpper in och ut laddade partiklar. När dessa portar fungerar felaktigt kan resultatet bli epilepsi hos nyfödda. Denna studie undersöker en sådan port, en kaliumkanal kallad KCNQ2, och avslöjar ett tidigare okänt sätt som cellen verkar klippa detta protein i bitar — en process som kan hjälpa till att förklara varför vissa förändringar i KCNQ2‑genen ger upphov till lindriga, kortvariga anfall medan andra leder till svåra, livslånga hjärnproblem.
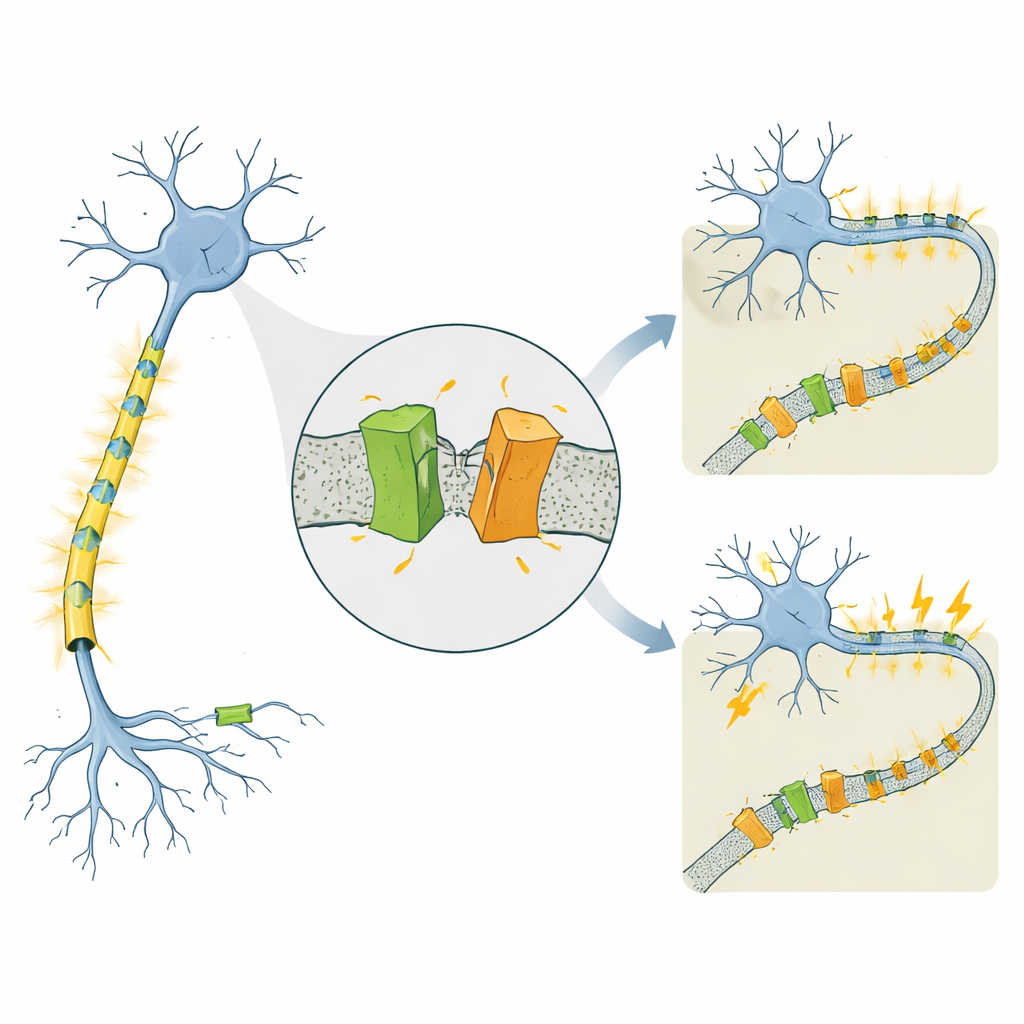
En kanal kopplad till mycket olika epilepsier
KCNQ2 tillhör en familj av spänningsstyrda kaliumkanaler som fungerar som bromsar för nervceller och hjälper dem att återställa sig efter varje elektrisk puls. Mutationer i den mänskliga KCNQ2‑genen är kända för att orsaka två slående olika tillstånd som båda börjar under de första levnadsdagarna. Vid självbegränsad familjär neonatal epilepsi (SLFNE) slutar anfallen vanligtvis inom veckor och barnen mår bra. Vid utvecklings‑ och epileptisk encefalopati (DEE) är anfallen svårare att kontrollera och åtföljs av allvarliga utvecklingsförseningar. Många sjukdomsframkallande KCNQ2‑mutationer hade katalogiserats, men det var oklart varför vissa förändringar leder till ett relativt gott förlopp medan andra ger svår funktionsnedsättning, särskilt eftersom den totala mängden KCNQ2‑protein ofta ser likartad ut.
En överraskande klyvning i kanalen
Forskarna studerade mus‑ och människoversioner av KCNQ2 med fokus på flera patient‑härledda mutationer. De märkte proteinet så att de kunde se både dess början och slut på laboratoriegeler. Som förväntat såg de den fullängds‑kanalen som sitter i cellmembranet. Men de upptäckte också konsekvent två mindre fragment: ett från proteinets framände och ett från dess svans. Båda fragmenten hittades i den membranrika delen av cellen, vilket indikerar att hela kanalen bildas först och sedan klipps, snarare än att korta former översätts från början. Det pekar på en tidigare oigenkänd posttranslationell beskärning — i praktiken cellens proteasax som skär KCNQ2 i två membranbundna fragment.
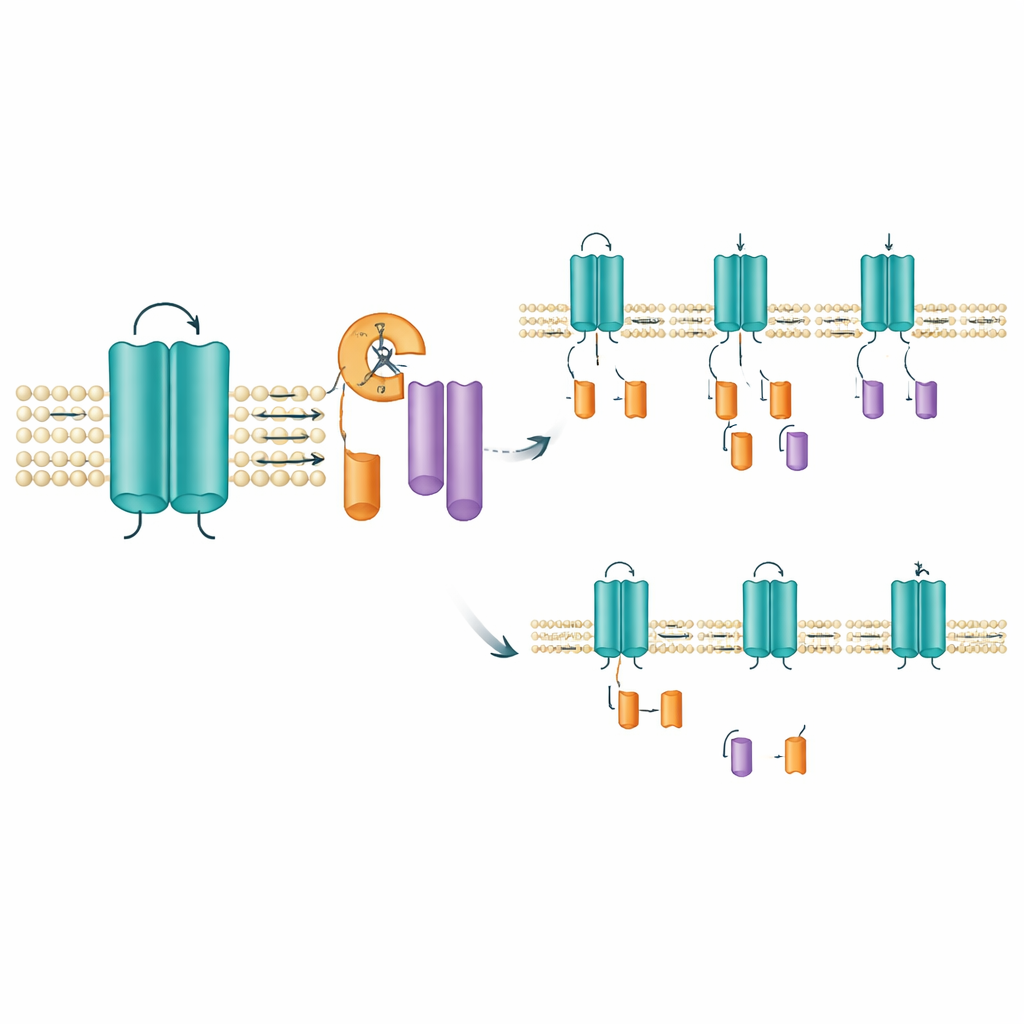
Att lokalisera var och hur klyvningen sker
För att hitta den sannolika klyvningsplatsen tog teamet bort små aminosyresträckor inom det område som omfattar ett av kanalens spänningskänsliga segment. Att ta bort ett smalt fönster om tio aminosyror (positionerna 171–180) gjorde att svansfragmentet försvann medan fullängds‑kanalen förblev intakt, vilket starkt tyder på att klyvningen sker i eller nära detta korta avsnitt. Detta område ligger inom ett membranigenomgående segment som hjälper kanalen att svara på spänningsförändringar, så beskärning här kan ändra hur kanalen beter sig eller hur länge den stannar på plats. Intressant nog förhindrade varken standardverktyg för att förutsäga var kända proteaser klyver proteiner, eller bredspektrum‑proteashämmare, bildandet av fragmenten. Det öppnar för att en specialiserad membraninkapslad enzymtyp, av den sort som redan är känd för att trimma andra signalproteiner, kan ligga bakom.
Mutationer rubbar fragmentbalansen
Inte alla KCNQ2‑mutationer påverkade fragmenten på samma sätt. I celler som uttryckte mutationer kopplade till den mildare SLFNE‑formen, såsom A306T och en viss förändring vid position 284 (Y284C), ökade mängden svansfragment jämfört med den normala kanalen. Däremot gav flera DEE‑associerade mutationer, inklusive en annan förändring på exakt samma position (Y284D), färre fragment. Dessa motsatta skiften — fler fragment i vissa varianter, färre i andra — uppstod trots att den totala mängden fullängds‑kanal var liknande. Samma mönster framträdde när teamet undersökte den mänskliga versionen av KCNQ2 och när de testade olika celltyper, inklusive mänskliga nervliknande celler. En närbesläktad kanal, KCNQ3, visade ingen sådan klyvning alls, vilket understryker att denna beskärning är en specifik, bevarad egenskap hos KCNQ2.
Vad detta betyder för epilepsi
Denna studie visar att KCNQ2 inte bara tillverkas och placeras i membranet som en enda statisk enhet. Istället klipper cellerna rutinmässigt kanalen i två membranankrade fragment genom en fortfarande gåtfull process, och sjukdomsframkallande mutationer skiftar hur mycket av kanalen som finns i fullängds‑ respektive klyvd form. Även om fragmentens exakta roll ännu inte är känd tyder deras bevarade närvaro över arter och celltyper på att de ingår i normal kanalreglering snarare än att vara rent experimentella artefakter. Subtila skillnader i hur effektivt KCNQ2 klyvs — och hur de resulterande bitarna påverkar kanalens aktivitet eller livslängd — kan bidra till att förklara varför vissa genetiska förändringar ger korta, självbegränsade anfall medan andra banar väg för svår, bestående epilepsi. Framtida studier som identifierar det ansvariga klyvningsenzymet och testar hur fragmenten påverkar kanalfunktionen kan öppna nya vägar för riktade behandlingar mot epilepsi.
Citering: Kimura, Y., Uchiyama, H., Masuda, K. et al. Novel proteolytic post-translational modification in voltage-gated potassium channel KCNQ2. Sci Rep 16, 11954 (2026). https://doi.org/10.1038/s41598-026-42444-9
Nyckelord: KCNQ2, kaliumkanal, epilepsi, posttranslationell modifiering, proteolytisk klyvning