Clear Sky Science · es
Nueva modificación postraduccional proteolítica en el canal de potasio dependiente de voltaje KCNQ2
Por qué importan estas pequeñas compuertas cerebrales
Nuestros pensamientos, movimientos e incluso el primer llanto de un recién nacido dependen de miles de millones de neuronas que disparan en patrones cuidadosamente sincronizados. Estas señales eléctricas están controladas por “compuertas” microscópicas en la membrana celular que permiten la entrada y salida de partículas cargadas. Cuando estas compuertas funcionan mal, el resultado puede ser epilepsia en neonatos. Este estudio investiga una de esas compuertas, un canal de potasio llamado KCNQ2, y descubre una forma hasta ahora desconocida en que la célula parece recortar esta proteína en trozos —un proceso que podría ayudar a explicar por qué algunos cambios en el gen KCNQ2 causan convulsiones leves y de corta duración mientras que otros conducen a problemas cerebrales graves y crónicos.
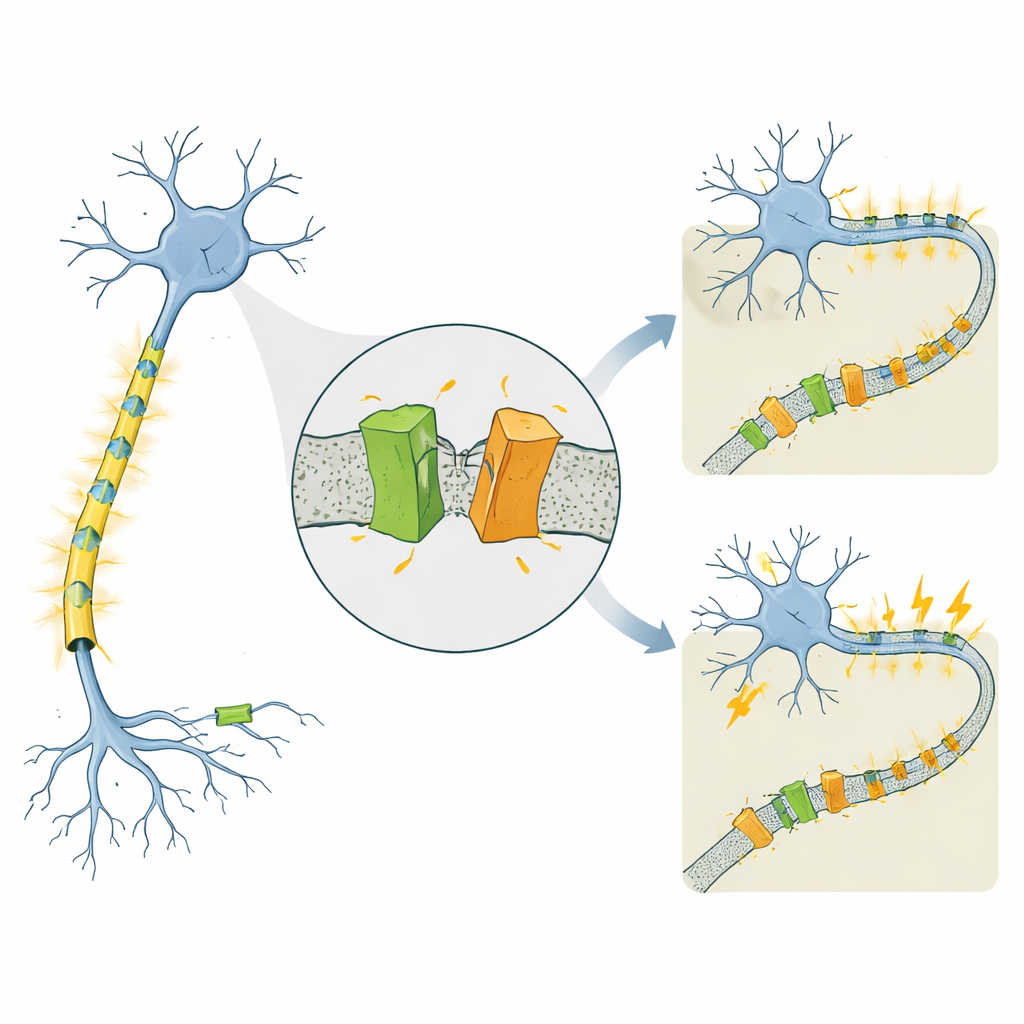
Un canal vinculado a epilepsias muy diferentes
KCNQ2 forma parte de una familia de canales de potasio dependientes de voltaje que actúan como frenos en las neuronas, ayudándolas a restablecerse tras cada pulso eléctrico. Se sabe que mutaciones en el gen humano KCNQ2 causan dos condiciones notablemente distintas que comienzan en los primeros días de vida. En la epilepsia neonatal familiar autolimitada (SLFNE), las convulsiones suelen cesar en semanas y los niños evolucionan bien. En la encefalopatía epiléptica y del desarrollo (DEE), las convulsiones son más difíciles de controlar y se acompañan de graves retrasos del desarrollo. Se habían catalogado muchas mutaciones patogénicas de KCNQ2, pero seguía sin estar claro por qué algunas variaciones conducen a un curso relativamente benigno mientras que otras producen una discapacidad severa, especialmente dado que los niveles totales de proteína KCNQ2 a menudo parecen similares.
Un corte sorprendente en el canal
Los investigadores estudiaron versiones de KCNQ2 de ratón y humano, centrándose en varias mutaciones derivadas de pacientes. Marcaban la proteína para poder ver tanto su extremo N como su extremo C en geles de laboratorio. Como era de esperar, observaron el canal de longitud completa, que se localiza en la membrana celular. Pero también detectaron de forma constante dos fragmentos más pequeños: uno correspondiente al extremo inicial de la proteína y otro a su cola. Ambos fragmentos se encontraron en la fracción rica en membranas de la célula, lo que indica que primero se sintetiza el canal completo y luego se corta, en lugar de ser traducido desde el inicio como formas cortas. Esto señalaba un evento de recorte postraduccional hasta ahora no reconocido—esencialmente, unas tijeras proteicas celulares que parten KCNQ2 en dos fragmentos anclados a la membrana.
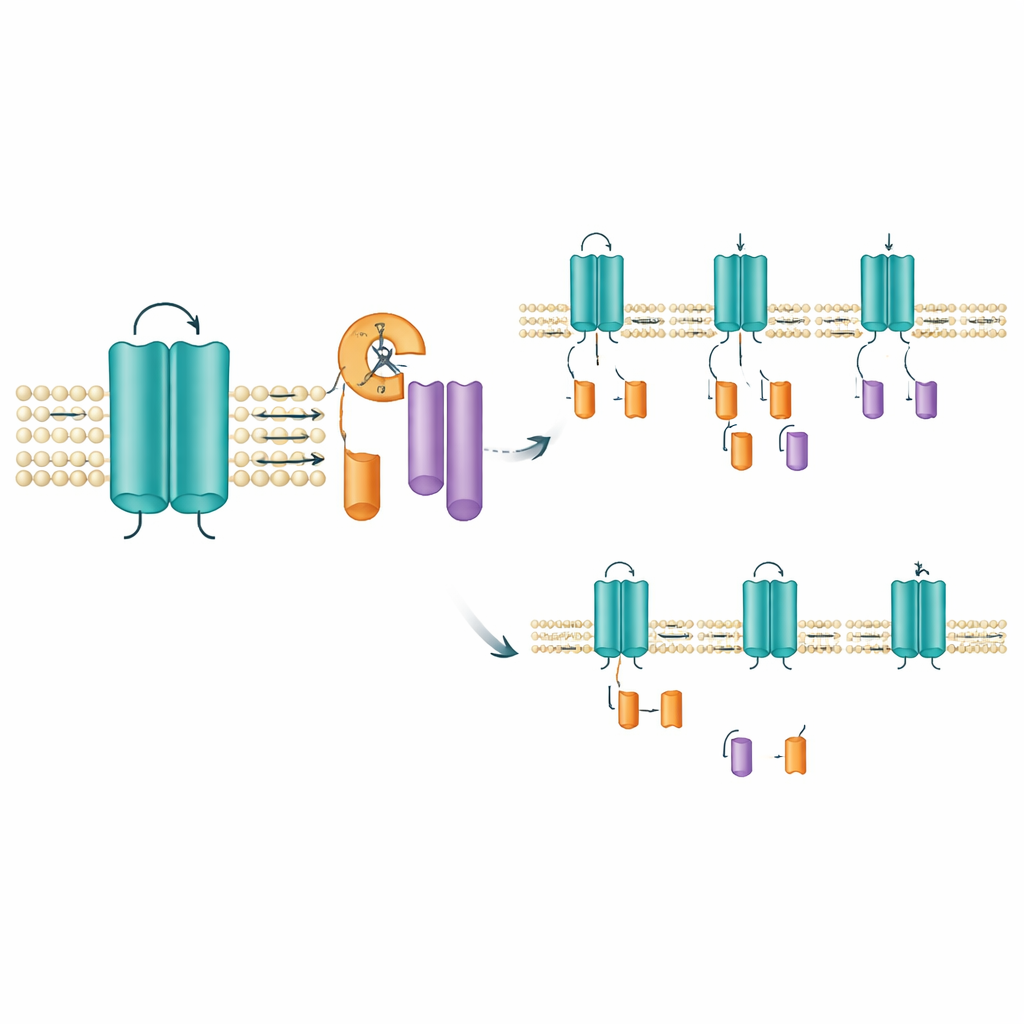
Localizando dónde y cómo ocurre el corte
Para identificar el probable sitio de corte, el equipo eliminó pequeños tramos de aminoácidos dentro de la región que abarca uno de los segmentos sensores de voltaje del canal. La eliminación de una ventana estrecha de diez aminoácidos (posiciones 171–180) abolió la aparición del fragmento de la cola mientras dejaba intacto el canal de longitud completa, lo que sugiere con fuerza que la escisión ocurre en o cerca de ese corto tramo. Esta zona se sitúa dentro de un segmento que atraviesa la membrana y ayuda al canal a responder a cambios de voltaje, por lo que un recorte ahí podría alterar el comportamiento del canal o su tiempo de permanencia en la membrana. De manera intrigante, las herramientas estándar para predecir dónde cortan las enzimas digestivas conocidas, así como bloqueadores de proteasas de amplio espectro, no impidieron la formación de los fragmentos. Eso plantea la posibilidad de que una enzima especializada embebida en la membrana, del tipo ya conocido por recortar otras proteínas de señalización, sea la responsable.
Las mutaciones inclinan el equilibrio de fragmentos
No todas las mutaciones de KCNQ2 afectaron los fragmentos de la misma manera. En células que expresaban mutaciones asociadas con la forma más leve SLFNE, como A306T y un cambio particular en la posición 284 (Y284C), la cantidad del fragmento de la cola aumentó en comparación con el canal normal. En contraste, varias mutaciones vinculadas a DEE, incluida una alteración distinta en la misma posición (Y284D), produjeron menos fragmentos. Estos desplazamientos opuestos —más fragmentos en algunas variantes, menos en otras— ocurrieron pese a que la cantidad total del canal de longitud completa era similar. El mismo patrón apareció cuando el equipo examinó la versión humana de KCNQ2 y cuando probaron distintos tipos celulares, incluidas células humanas con rasgos neuronales. Un canal relacionado, KCNQ3, no mostró este corte en absoluto, lo que subraya que este recorte es una característica específica y conservada de KCNQ2.
Qué significa esto para la epilepsia
Este trabajo revela que KCNQ2 no se fabrica y sitúa en la membrana como una sola pieza estática. En cambio, las células rutinariamente lo recortan en dos fragmentos anclados a la membrana mediante un proceso aún misterioso, y las mutaciones que causan enfermedad modifican la proporción entre canal íntegro y canal recortado. Aunque el papel exacto de estos fragmentos aún se desconoce, su conservación entre especies y tipos celulares sugiere que forman parte del control normal del canal y no son meros artefactos de laboratorio. Diferencias sutiles en la eficiencia del recorte de KCNQ2 —y en cómo las piezas resultantes influyen en la actividad o la vida útil del canal— podrían ayudar a explicar por qué algunos cambios genéticos provocan convulsiones breves y autolimitadas mientras que otros preparan el terreno para una epilepsia severa y persistente. Estudios futuros que identifiquen la enzima responsable del corte y prueben cómo afectan los fragmentos a la función del canal podrían abrir nuevos caminos para tratamientos dirigidos de la epilepsia.
Cita: Kimura, Y., Uchiyama, H., Masuda, K. et al. Novel proteolytic post-translational modification in voltage-gated potassium channel KCNQ2. Sci Rep 16, 11954 (2026). https://doi.org/10.1038/s41598-026-42444-9
Palabras clave: KCNQ2, canal de potasio, epilepsia, modificación postraduccional, corte proteolítico