Clear Sky Science · en
Novel proteolytic post-translational modification in voltage-gated potassium channel KCNQ2
Why tiny brain gates matter
Our thoughts, movements, and even newborns’ first cries depend on billions of nerve cells firing in carefully timed patterns. These electrical signals are controlled by microscopic “gates” in the cell membrane that let charged particles in and out. When these gates malfunction, the result can be epilepsy in newborn babies. This study looks at one such gate, a potassium channel called KCNQ2, and uncovers a previously unknown way the cell appears to clip this protein into pieces—a process that may help explain why some KCNQ2 gene changes cause mild, short‑lived seizures while others lead to severe, lifelong brain problems.
A channel linked to very different epilepsies
KCNQ2 is part of a family of voltage‑gated potassium channels that act like brakes on nerve cells, helping them reset after each electrical pulse. Mutations in the human KCNQ2 gene are known to cause two strikingly different conditions that both begin in the first days of life. In self‑limited familial neonatal epilepsy (SLFNE), seizures usually stop within weeks and children do well. In developmental and epileptic encephalopathy (DEE), seizures are harder to control and are accompanied by serious developmental delays. Many disease‑causing KCNQ2 mutations had been cataloged, but it remained unclear why some changes lead to a relatively benign course while others produce severe disability, especially since overall levels of the KCNQ2 protein often look similar.
A surprising cut in the channel
The researchers studied mouse and human versions of KCNQ2, focusing on several patient‑derived mutations. They tagged the protein so they could see both its beginning and end on laboratory gels. As expected, they saw the full‑length channel, which sits in the cell membrane. But they also consistently detected two smaller pieces: one from the protein’s front end and one from its tail. Both fragments were found in the membrane‑rich fraction of the cell, indicating that the complete channel is made first and then cut, rather than being translated as short forms from the start. This pointed to a previously unrecognized “post‑translational” trimming event—essentially, the cell’s protein scissors slicing KCNQ2 into two membrane‑bound fragments.
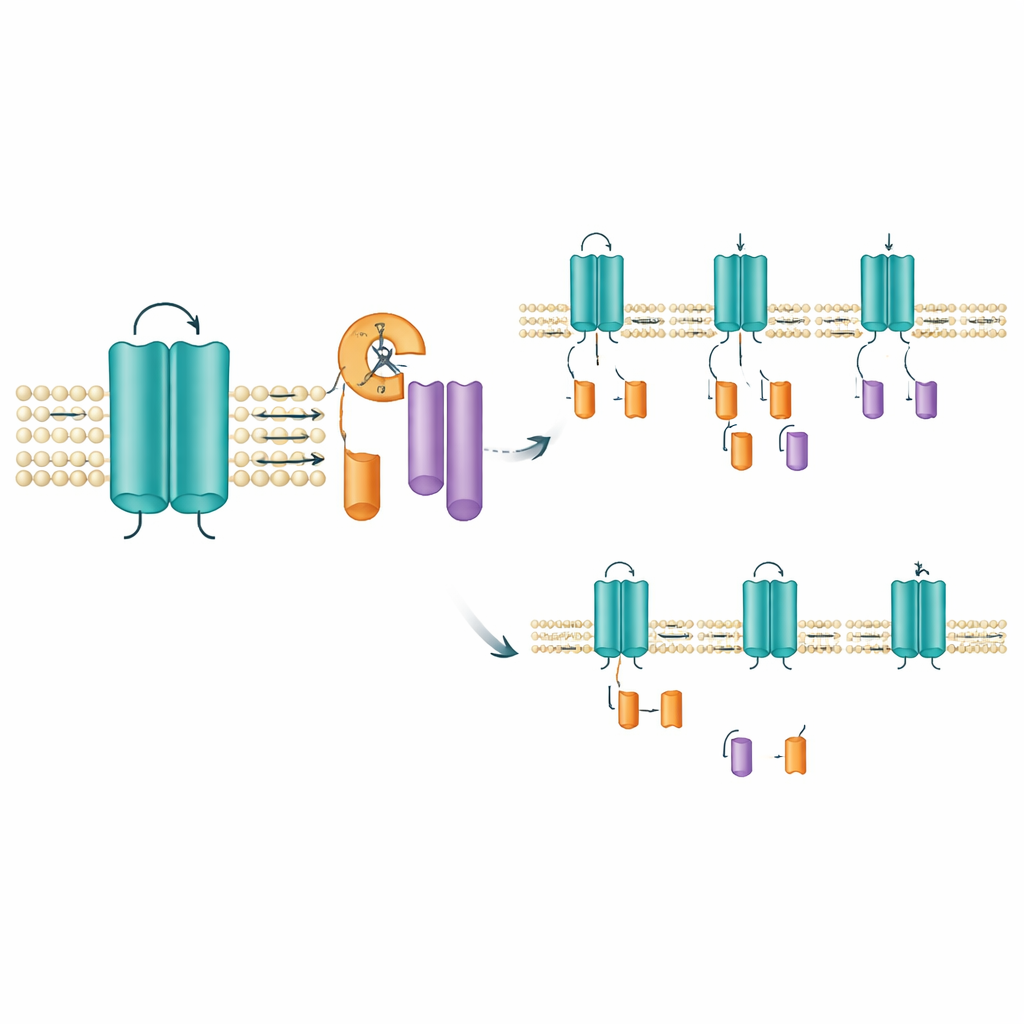
Pinpointing where and how the cut happens
To find the likely cutting site, the team deleted small stretches of amino acids within the region that spans one of the channel’s voltage‑sensing segments. Removing a narrow window of ten amino acids (positions 171–180) abolished the appearance of the tail fragment while leaving the full‑length channel intact, strongly suggesting that the cleavage occurs in or near this short stretch. This area lies within a membrane‑spanning segment that helps the channel respond to voltage changes, so clipping here could change how the channel behaves or how long it stays in place. Intriguingly, standard tools for predicting where known digestive enzymes cut proteins, as well as broad‑spectrum protease blockers, did not prevent fragment formation. That raises the possibility that a specialized membrane‑embedded enzyme, of the kind already known to trim other signaling proteins, may be responsible.
Mutations tilt the balance of fragments
Not all KCNQ2 mutations affected the fragments in the same way. In cells expressing mutations tied to the milder SLFNE form, such as A306T and a particular change at position 284 (Y284C), the amount of tail fragment increased compared with the normal channel. By contrast, several DEE‑linked mutations, including a different change at the very same position (Y284D), produced fewer fragments. These opposite shifts—more fragments in some variants, fewer in others—occurred even though the total amount of full‑length channel was similar. The same pattern appeared when the team examined the human version of KCNQ2 and when they tested different cell types, including human nerve‑like cells. A related channel, KCNQ3, did not show this cleavage at all, underscoring that this trimming is a specific, conserved feature of KCNQ2.
What this means for epilepsy
This work reveals that KCNQ2 is not simply made and placed in the membrane as a single, static piece. Instead, cells routinely clip it into two membrane‑anchored fragments through a still‑mysterious process, and disease‑causing mutations shift how much of the channel ends up in full‑length versus cut form. Although the exact role of these fragments is not yet known, their conservation across species and cell types suggests they are part of normal channel control rather than mere laboratory artifacts. Subtle differences in how efficiently KCNQ2 is cleaved—and how the resulting pieces influence channel activity or lifespan—may help explain why some genetic changes lead to brief, self‑limited seizures while others set the stage for severe, persistent epilepsy. Future studies that identify the responsible cutting enzyme and test how the fragments affect channel function could open new paths for targeted epilepsy treatments.
Citation: Kimura, Y., Uchiyama, H., Masuda, K. et al. Novel proteolytic post-translational modification in voltage-gated potassium channel KCNQ2. Sci Rep 16, 11954 (2026). https://doi.org/10.1038/s41598-026-42444-9
Keywords: KCNQ2, potassium channel, epilepsy, post-translational modification, proteolytic cleavage