Clear Sky Science · de
Neuartige proteolytische posttranslationale Modifikation im spannungsabhängigen Kaliumkanal KCNQ2
Warum winzige Hirn‑Tore wichtig sind
Unsere Gedanken, Bewegungen und sogar der erste Schrei Neugeborener beruhen auf Milliarden von Nervenzellen, die in zeitlich präzisen Mustern feuern. Diese elektrischen Signale werden von mikroskopischen „Toren“ in der Zellmembran gesteuert, die geladene Teilchen hinein- und hinauslassen. Wenn diese Tore nicht richtig funktionieren, kann das bei Neugeborenen zu Epilepsie führen. Diese Studie betrachtet eines dieser Tore, einen Kaliumkanal namens KCNQ2, und enthüllt eine bisher unbekannte Art, wie die Zelle dieses Protein offenbar in Stücke schneidet — ein Prozess, der helfen könnte zu erklären, warum einige Veränderungen im KCNQ2‑Gen zu milden, kurzlebigen Anfällen führen, während andere schwere, lebenslange Hirnprobleme verursachen.
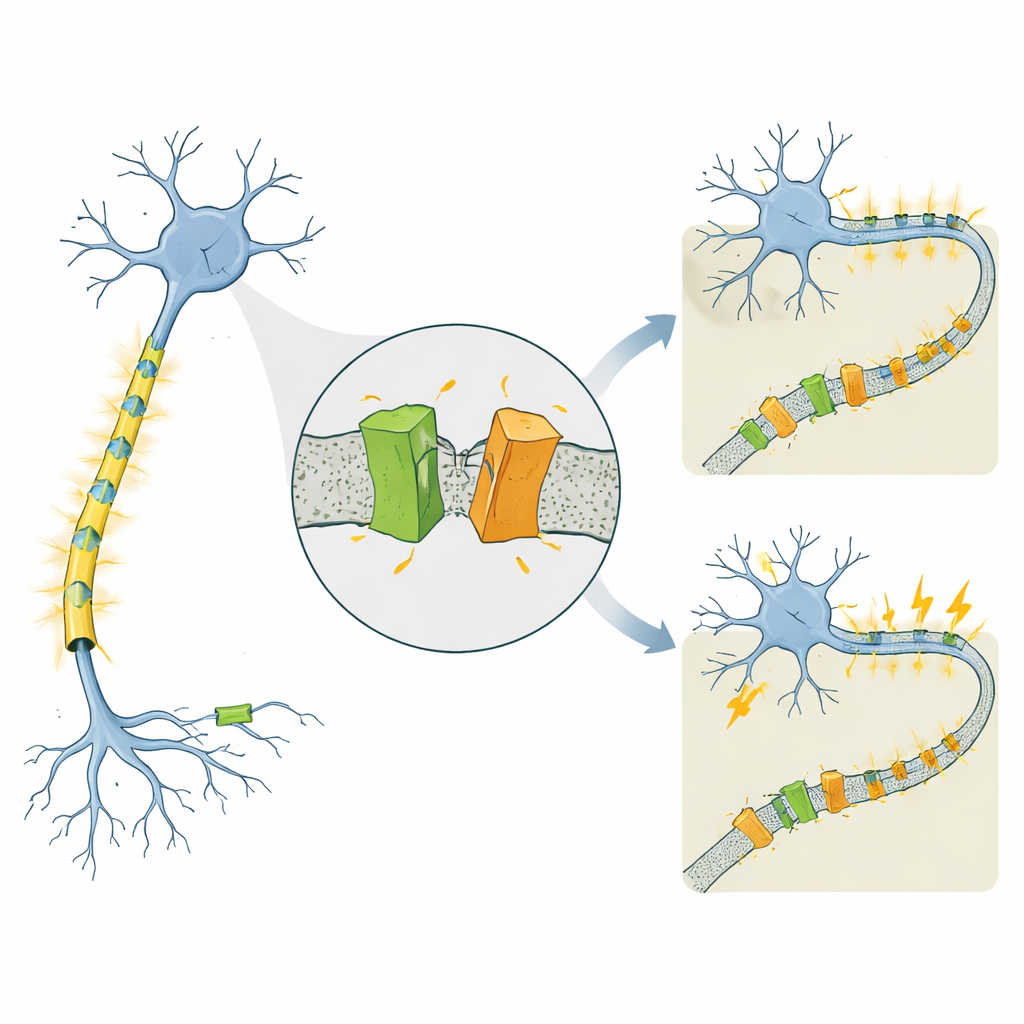
Ein Kanal, der mit sehr verschiedenen Epilepsien verknüpft ist
KCNQ2 gehört zu einer Familie spannungsabhängiger Kaliumkanäle, die wie Bremsen für Nervenzellen wirken und ihnen helfen, sich nach jedem elektrischen Impuls zurückzusetzen. Mutationen im menschlichen KCNQ2‑Gen sind dafür bekannt, zwei auffallend unterschiedliche Zustände hervorzurufen, die beide in den ersten Lebenstagen beginnen. Bei der selbstbegrenzten familiären neonatalen Epilepsie (SLFNE) hören die Anfälle meist innerhalb von Wochen auf und die Kinder entwickeln sich gut. Bei der entwicklungsbedingten und epileptischen Enzephalopathie (DEE) sind die Anfälle schwerer zu kontrollieren und gehen mit erheblichen Entwicklungsverzögerungen einher. Viele krankheitsverursachende KCNQ2‑Mutationen waren bereits katalogisiert, doch blieb unklar, warum einige Veränderungen einen relativ milden Verlauf verursachen, während andere zu schwerer Behinderung führen — insbesondere da die Gesamtmengen des KCNQ2‑Proteins häufig ähnlich aussehen.
Ein überraschender Schnitt im Kanal
Die Forschenden untersuchten Maus‑ und Humanversionen von KCNQ2 und konzentrierten sich auf mehrere patientenabgeleitete Mutationen. Sie markierten das Protein, um sowohl dessen Anfangs‑ als auch Endabschnitt in Laborgelen sichtbar zu machen. Wie erwartet sahen sie den volllängigen Kanal, der in der Zellmembran sitzt. Sie entdeckten aber auch konsequent zwei kleinere Fragmente: eines vom N‑Terminus und eines vom C‑Terminus des Proteins. Beide Fragmente fanden sich in der membranreichen Fraktion der Zelle, was darauf hindeutet, dass der vollständige Kanal zunächst hergestellt und dann geschnitten wird, statt von Anfang an als kurze Formen translatiert zu werden. Das deutet auf ein bisher unerkanntes posttranslationales Beschnitt‑Ereignis hin — im Grunde die Proteinschere der Zelle, die KCNQ2 in zwei membrangebundenen Fragmenten zerteilt.
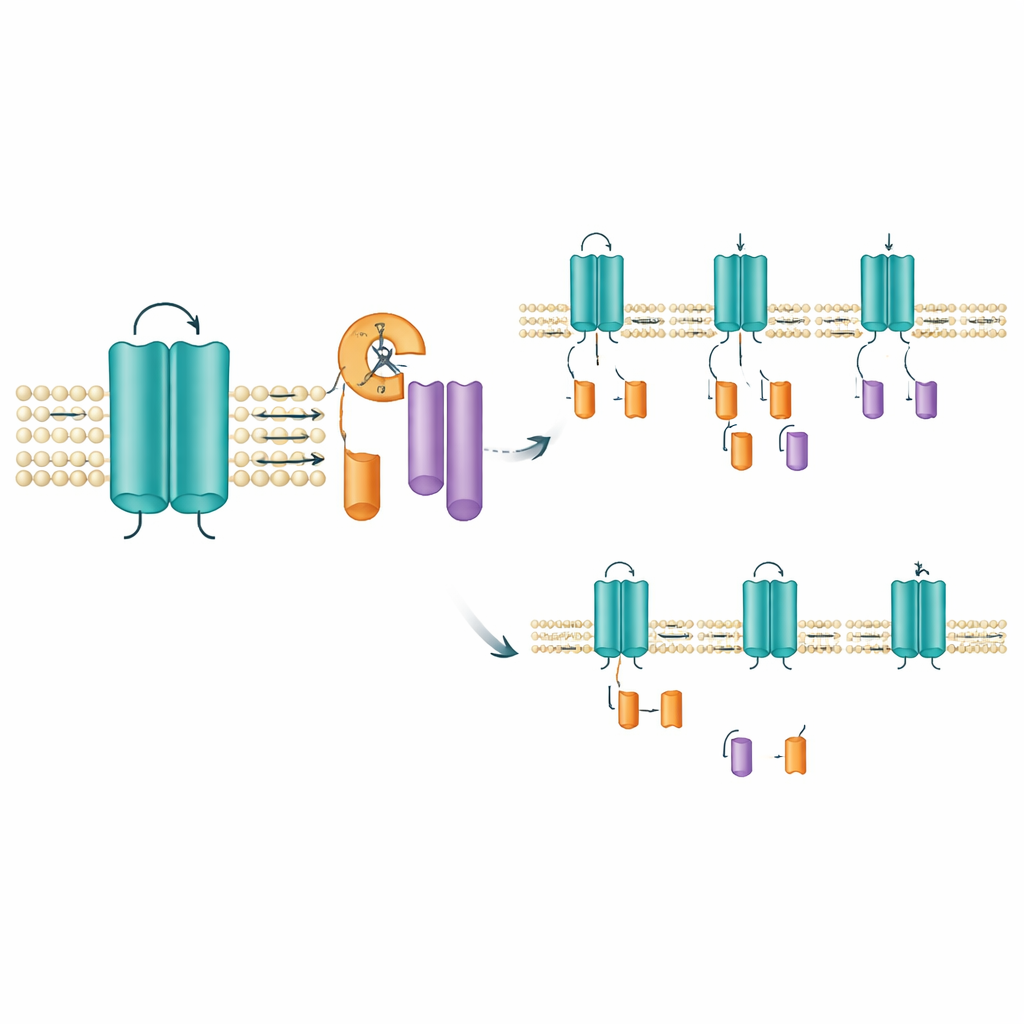
Lokalisierung, wo und wie der Schnitt stattfindet
Um die wahrscheinliche Schnittstelle zu finden, löschte das Team kleine Aminosäure‑Abschnitte innerhalb der Region, die eines der spannungssensorischen Segmente des Kanals überspannt. Das Entfernen eines schmalen Fensters von zehn Aminosäuren (Positionen 171–180) beseitigte das Auftreten des C‑Terminal‑Fragments, während der volllängige Kanal erhalten blieb — ein starker Hinweis darauf, dass die Spaltung in oder nahe diesem kurzen Abschnitt stattfindet. Dieses Gebiet liegt innerhalb eines membranüberspannenden Segments, das dem Kanal hilft, auf Spannungsänderungen zu reagieren, sodass ein Schnitt hier das Verhalten des Kanals oder seine Verweildauer in der Membran verändern könnte. Interessanterweise verhinderten Standardwerkzeuge zur Vorhersage bekannter Proteasen‑Schnittstellen sowie Breitband‑Proteasehemmer die Fragmentbildung nicht. Das legt die Möglichkeit nahe, dass eine spezialisierte, membranverankerte Enzymklasse, wie sie bereits bekannt ist, um andere Signalmoleküle zu trimmen, dafür verantwortlich sein könnte.
Mutationen verschieben das Gleichgewicht der Fragmente
Nicht alle KCNQ2‑Mutationen beeinflussten die Fragmentbildung auf dieselbe Weise. In Zellen, die Mutationen exprimierten, die mit der milderen SLFNE‑Form verknüpft sind, wie A306T und eine bestimmte Veränderung an Position 284 (Y284C), nahm die Menge des C‑Terminal‑Fragments im Vergleich zum normalen Kanal zu. Dagegen erzeugten mehrere DEE‑assoziierte Mutationen, einschließlich einer anderen Veränderung an derselben Position (Y284D), weniger Fragmente. Diese entgegengesetzten Verschiebungen — mehr Fragmente bei einigen Varianten, weniger bei anderen — traten auf, obwohl die Gesamtmenge des volllängigen Kanals ähnlich war. Dasselbe Muster zeigte sich bei Untersuchungen der humanen KCNQ2‑Variante und in verschiedenen Zelltypen, einschließlich humaner, nervenzellähnlicher Zellen. Ein verwandter Kanal, KCNQ3, zeigte diese Spaltung überhaupt nicht, was unterstreicht, dass dieses Trimmen eine spezifische, für KCNQ2 konservierte Eigenschaft ist.
Was das für Epilepsien bedeutet
Diese Arbeit zeigt, dass KCNQ2 nicht einfach als ein einziges, statisches Stück hergestellt und in die Membran eingebaut wird. Stattdessen schneiden Zellen es routinemäßig in zwei membranverankerte Fragmente durch einen noch unerforschten Prozess, und krankheitsverursachende Mutationen verschieben, wie viel des Kanals in Vollform gegenüber der geschnittenen Form vorkommt. Obwohl die genaue Funktion dieser Fragmente noch nicht bekannt ist, spricht ihre Erhaltung über Arten und Zelltypen hinweg dafür, dass sie Teil der normalen Kanalregulation und keine bloßen Laborartefakte sind. Feine Unterschiede in der Effizienz der KCNQ2‑Spaltung — und wie die entstehenden Stücke die Kanalaktivität oder Lebensdauer beeinflussen — könnten erklären, warum einige genetische Veränderungen zu kurzen, selbstbegrenzten Anfällen führen, während andere den Grundstein für schwere, anhaltende Epilepsien legen. Zukünftige Studien, die das verantwortliche Schneideenzym identifizieren und testen, wie die Fragmente die Kanal‑Funktion beeinflussen, könnten neue Wege für gezielte Epilepsiebehandlungen eröffnen.
Zitation: Kimura, Y., Uchiyama, H., Masuda, K. et al. Novel proteolytic post-translational modification in voltage-gated potassium channel KCNQ2. Sci Rep 16, 11954 (2026). https://doi.org/10.1038/s41598-026-42444-9
Schlüsselwörter: KCNQ2, Kaliumkanal, Epilepsie, posttranslationale Modifikation, proteolytische Spaltung