Clear Sky Science · fr
Nouvelle modification post‑traductionnelle protéolytique dans le canal potassique voltage‑dépendant KCNQ2
Pourquoi ces minuscules portes cérébrales comptent
Nos pensées, nos mouvements et même les premiers pleurs des nouveau‑nés dépendent de milliards de neurones qui s’activent selon des rythmes finement réglés. Ces signaux électriques sont contrôlés par de microscopiques « portes » dans la membrane cellulaire qui laissent entrer ou sortir des particules chargées. Quand ces portes dysfonctionnent, le résultat peut être une épilepsie chez le nouveau‑né. Cette étude porte sur l’une de ces portes, un canal potassique nommé KCNQ2, et révèle un mode jusque‑là inconnu par lequel la cellule semble découper cette protéine en morceaux — un processus qui pourrait aider à expliquer pourquoi certaines variantes du gène KCNQ2 provoquent des crises brèves et bénignes tandis que d’autres entraînent des troubles neurologiques sévères et durables.
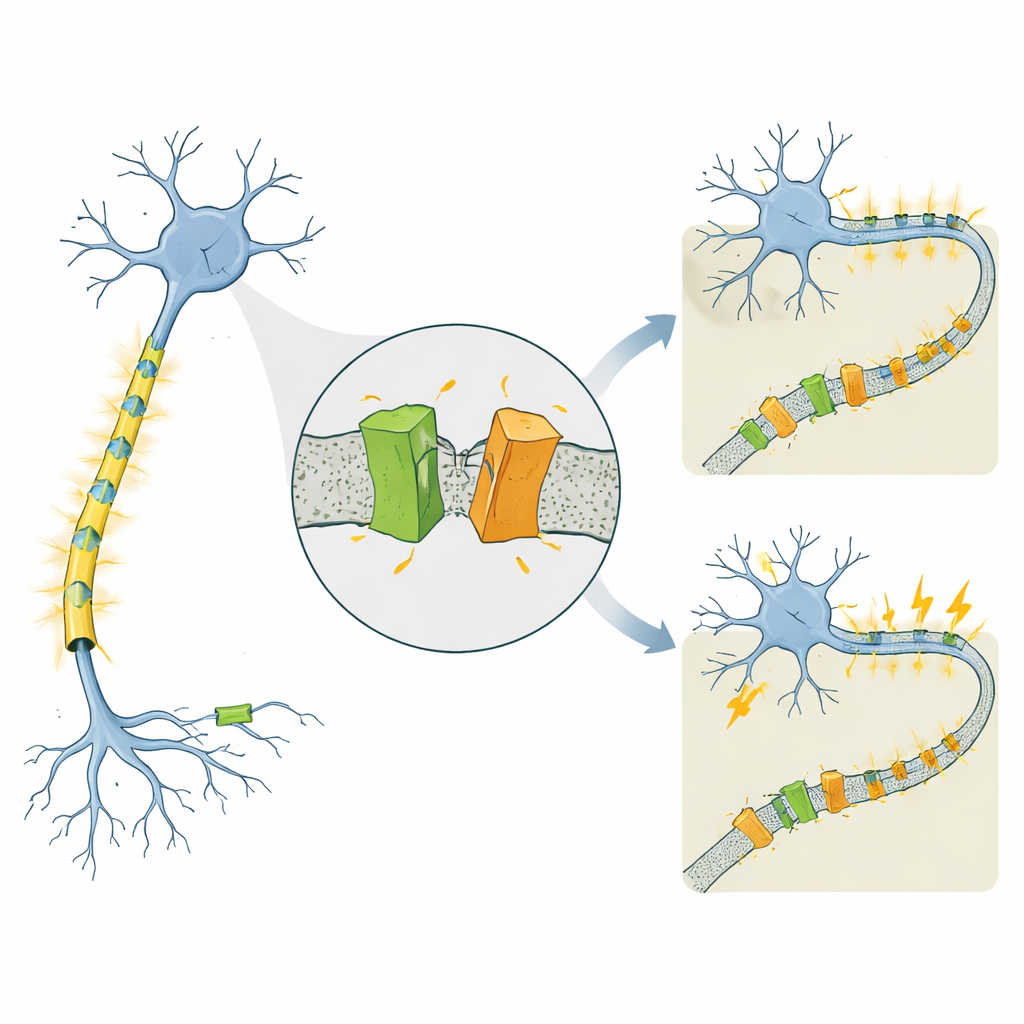
Un canal lié à des épilepsies très différentes
KCNQ2 fait partie d’une famille de canaux potassiques voltage‑dépendants qui jouent le rôle de freins sur les neurones, les aidant à se rétablir après chaque impulsion électrique. Des mutations du gène humain KCNQ2 sont connues pour provoquer deux affections nettement différentes qui débutent toutes deux dans les premiers jours de la vie. Dans l’épilepsie néonatale familiale à évolution favorable (self‑limited familial neonatal epilepsy, SLFNE), les crises cessent généralement en quelques semaines et les enfants se développent bien. Dans l’encéphalopathie développementale et épileptique (developmental and epileptic encephalopathy, DEE), les crises sont plus difficiles à contrôler et s’accompagnent de retards de développement importants. De nombreuses mutations pathogènes de KCNQ2 avaient été répertoriées, mais il restait difficile d’expliquer pourquoi certaines altérations conduisent à une évolution relativement bénigne alors que d’autres provoquent un handicap sévère, d’autant plus que les niveaux totaux de protéine KCNQ2 paraissent souvent similaires.
Une coupure surprenante dans le canal
Les chercheurs ont étudié les versions murine et humaine de KCNQ2, en se concentrant sur plusieurs mutations issues de patients. Ils ont marqué la protéine afin de visualiser à la fois son extrémité N‑terminale et son extrémité C‑terminale sur des gels de laboratoire. Comme prévu, ils ont observé la forme pleine longueur du canal, qui s’insère dans la membrane cellulaire. Mais ils ont aussi détecté de façon consistante deux fragments plus petits : l’un provenant de l’extrémité avant de la protéine et l’autre de son extrémité arrière. Les deux fragments se trouvaient dans la fraction riche en membranes de la cellule, ce qui indique que le canal complet est d’abord produit puis découpé, plutôt que traduit dès le départ sous forme tronquée. Cela pointe vers un événement de « trimming » post‑traductionnel jusque‑là méconnu — essentiellement, des ciseaux protéiques cellulaires qui scindent KCNQ2 en deux fragments ancrés dans la membrane.
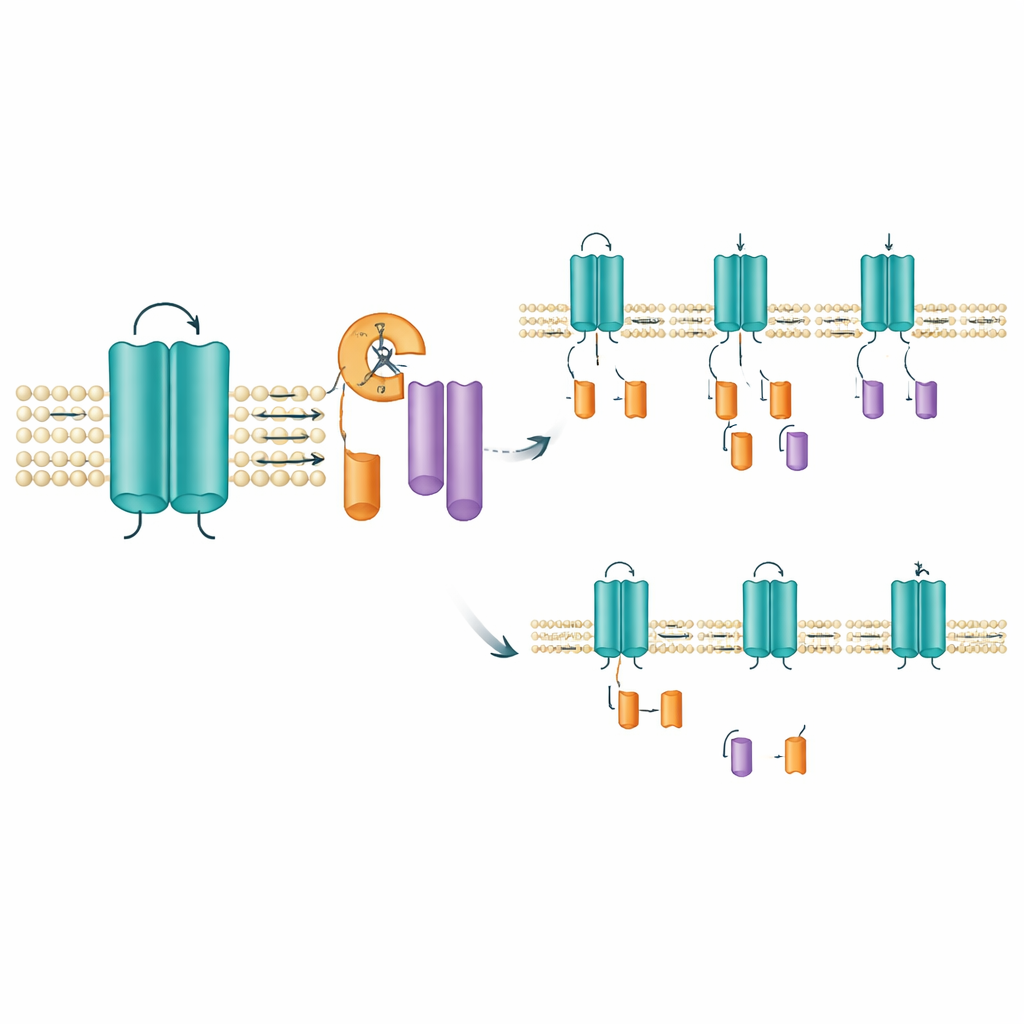
Localiser où et comment se produit la coupure
Pour identifier le site probable de clivage, l’équipe a supprimé de courts segments d’acides aminés dans la région qui traverse l’un des segments senseurs de tension du canal. La suppression d’une fenêtre étroite de dix acides aminés (positions 171–180) a fait disparaître l’apparition du fragment de queue tout en laissant le canal pleine longueur intact, suggérant fortement que le clivage se produit dans ou à proximité de cette courte séquence. Cette zone se situe dans un segment transmembranaire qui aide le canal à répondre aux variations de tension, de sorte qu’un clivage ici pourrait modifier le comportement du canal ou la durée pendant laquelle il reste en place. Fait intrigant, les outils habituels de prédiction des sites de coupure par des enzymes connues, ainsi que des inhibiteurs de protéases à large spectre, n’ont pas empêché la formation des fragments. Cela laisse ouverte la possibilité qu’une enzyme spécialisée intégrée à la membrane, du type déjà connu pour élaguer d’autres protéines de signalisation, soit responsable.
Les mutations déséquilibrent la balance des fragments
Toutes les mutations de KCNQ2 n’affectaient pas les fragments de la même manière. Dans des cellules exprimant des mutations associées à la forme plus bénigne SLFNE, comme A306T et une modification particulière en position 284 (Y284C), la quantité de fragment de la queue augmentait comparée au canal normal. En revanche, plusieurs mutations liées à la DEE, y compris une autre modification à cette même position (Y284D), entraînaient une diminution des fragments. Ces variations opposées — plus de fragments pour certaines variantes, moins pour d’autres — se produisaient alors que la quantité totale de canal pleine longueur restait similaire. Le même schéma est apparu lors de l’examen de la version humaine de KCNQ2 et dans différents types cellulaires, y compris des cellules d’aspect neuronal humaines. Un canal apparenté, KCNQ3, n’a montré aucun clivage, soulignant que ce trimming est une caractéristique spécifique et conservée de KCNQ2.
Quelles implications pour l’épilepsie
Ce travail révèle que KCNQ2 n’est pas simplement fabriqué et inséré dans la membrane sous une forme unique et statique. Au contraire, la cellule le découpe couramment en deux fragments ancrés à la membrane par un processus encore mystérieux, et les mutations pathogènes modulent la proportion de canal pleine longueur par rapport à la forme clivée. Bien que le rôle exact de ces fragments demeure inconnu, leur conservation entre espèces et types cellulaires suggère qu’ils participent au contrôle normal du canal plutôt qu’à de simples artefacts de laboratoire. Des différences subtiles dans l’efficacité du clivage de KCNQ2 — et dans la manière dont les fragments résultants influent sur l’activité ou la durée de vie du canal — pourraient contribuer à expliquer pourquoi certaines altérations génétiques conduisent à des crises brèves et auto‑limitantes tandis que d’autres prédisposent à une épilepsie sévère et persistante. Des études futures visant à identifier l’enzyme responsable du clivage et à tester l’impact des fragments sur la fonction du canal pourraient ouvrir de nouvelles voies pour des traitements ciblés de l’épilepsie.
Citation: Kimura, Y., Uchiyama, H., Masuda, K. et al. Novel proteolytic post-translational modification in voltage-gated potassium channel KCNQ2. Sci Rep 16, 11954 (2026). https://doi.org/10.1038/s41598-026-42444-9
Mots-clés: KCNQ2, canal potassique, épilepsie, modification post‑traductionnelle, clivage protéolytique