Clear Sky Science · sv
KAN-förstärkt kontrastiv inlärning: accelereraren för identifiering av kristallstrukturer från XRD-mönster
Varför snabbare kartläggning av kristaller spelar roll
Nya material för batterier, elektronik och ren energi upptäcks ofta en kristall i taget. Varje kristalls interna atomarrangemang avgör dess egenskaper, och forskare brukar läsa av detta arrangemang från pulverröntgendiffraktions (XRD) mönster—spetsiga diagram som uppstår när röntgenstrålar sprids i ett prov. I dag är omvandlingen av dessa mönster till en konkret struktur långsam och kräver experter. Denna artikel presenterar ett maskininlärningssystem som snabbt kan matcha ett XRD-mönster med troliga kristallstrukturer, vilket gör detektivarbetet snabbare, mer tillförlitligt och lätt att integrera i automatiserade laboratorier.
Från spetsiga mönster till atomära ritningar
I konventionell praxis undersöker en XRD-specialist ett mönsters toppar, använder fysikaliska formler för att härleda tänkbara atomavstånd och jämför därefter iterativt kandidatstrukturer mot datan. Denna process har svårt när toppar överlappar eller när det finns många snarlika alternativ, och den skalas inte väl till moderna höggenomströmningsexperiment som kan generera tusentals mönster per dag. Tidigare maskininlärningsverktyg har mest behandlat XRD som ett etiketteringsproblem—att förutsäga en symmetriklass eller rymdgrupp från ett mönster—instead of direkt identifiering av själva strukturen. Den nya metoden, kallad XRD‑Crystal Contrastive Pretraining (XCCP), omformulerar uppgiften som återhämtning: givet ett mönster, hitta den mest kompatibla kristallen i en stor databas.
En tvåögd syn på röntgenmönster
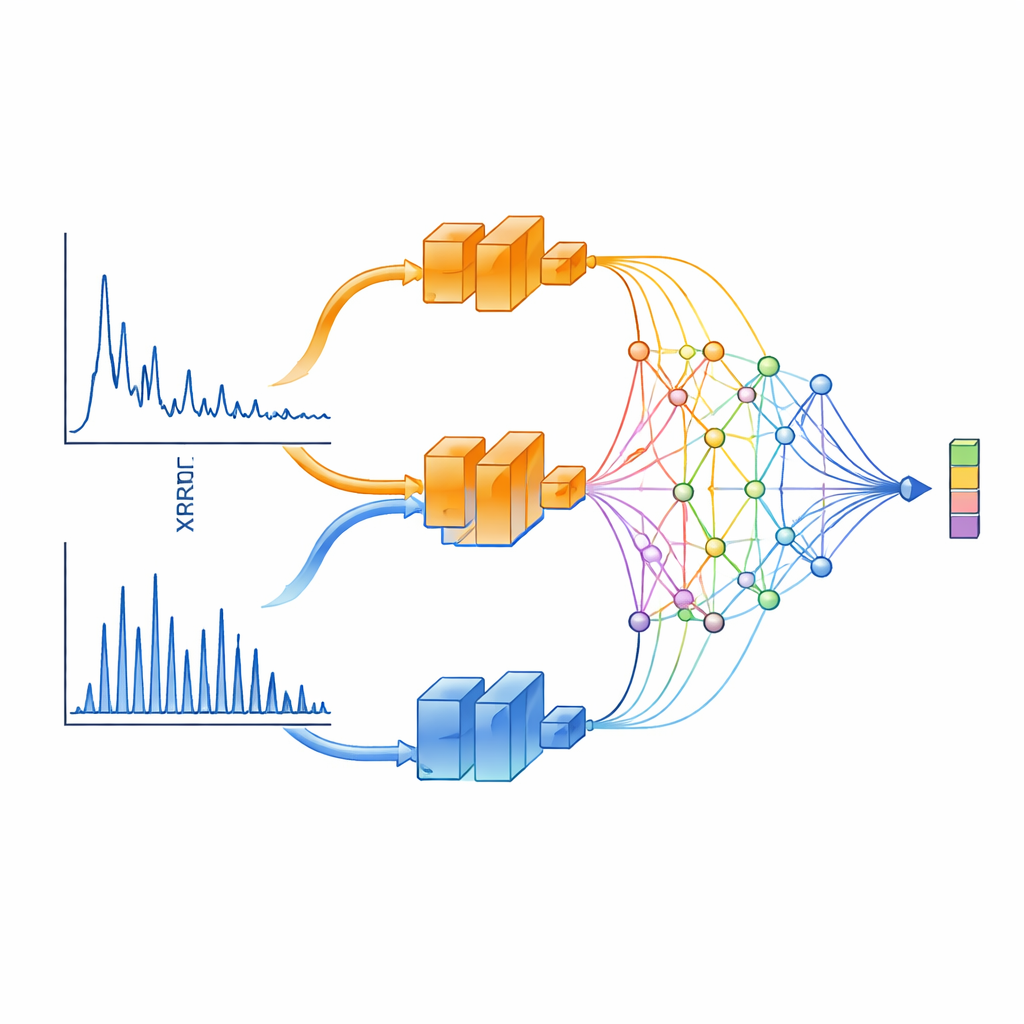
XCCP lär sig att ”se” XRD-data på ett fysikaliskt informerat sätt. Istället för att mata in hela mönstret i ett enda neuralt nätverk delar metoden upp det i två intervall. Den ena grenen fokuserar på små vinklar, som fångar långräckande egenskaper som skiktavstånd och supergitter. Den andra koncentrerar sig på stora vinklar, där topparna är täta och starkt styrda av kristallsymmetri. Varje gren bearbetas av ett djupt nätverk och kombineras sedan av en speciell projiceringsmodul baserad på Kolmogorov–Arnold-nätverk (KAN). Denna modul är mycket skicklig på att rikta uppmärksamhet mot smala regioner i mönstret—precis där skarpa diffraktionstoppar bär den mest strukturella informationen.
Låta mönster och strukturer mötas i mitten
På kristallsidan använder XCCP ett grafbaserat nätverk som representerar atomer som noder och deras bindningar som förbindelser. Under träningen ser systemet många matchade par: ett XRD-mönster och dess kända kristallstruktur. Det lär sig ett delat numeriskt rum där varje mönster ligger nära sin egen struktur och långt från felmatchade. När ett nytt mönster kommer in bäddar modellen in det i detta rum, jämför det med inbäddningar av alla databaskristaller och returnerar en rankad kortlista. Utan någon kunskap om vilka grundämnen som finns är korrekt struktur rankad som etta nästan hälften av gångerna och finns bland de fem översta i en stor majoritet av fallen. När användaren dessutom anger kemisk sammansättning—information som vanligtvis finns i verkliga experiment—är topp‑1-matchningen korrekt nästan 90 % av gångerna.
Se vad maskinen ser
Författarna undersöker om deras system förlitar sig på riktig fysik eller på slumpmässiga datakvibblor. Genom att maskera delar av mönstret och använda attribueringsverktyg visar de att KAN-huvudet baserar sina beslut främst på starka, välavgränsade diffraktionstoppar snarare än på breda bakgrundsvariationer eller brus. Den tillagda låg‑vinkelgrenen förbättrar konsekvent prestandan, särskilt för låg‑symmetriska kristaller och mönster där högvinkel‑funktioner är tvetydiga. Modellen visar sig också robust mot vanliga experimentella ofullkomligheter såsom toppbredning och små skift längs vinkelaxeln, och den överförs hyfsat väl till verkliga experimentella dataset. Viktigt är att de likhetsmått den producerar också fungerar som förtroendemått, som sjunker markant när den verkliga strukturen saknas i databasen—en väsentlig egenskap för säker användning i verkliga tillämpningar.
Mot smartare, självkörande materialupptäckt
För en icke-specialist är huvudbudskapet att XCCP förvandlar XRD‑analys från ett hantverk till en snabb, datadriven sökning. Genom att anpassa diffraktionsmönster och kandidatkristaller i ett delat rum, och genom att använda fysikmedveten nätverksdesign, kan systemet snabbt föreslå en kort lista med realistiska atomära ritningar med tolkbar säkerhet. Det ersätter inte expertbedömning eller detaljerad raffinering, men det accelererar i hög grad det första, svåraste steget—att avgöra vilka strukturer som överhuvudtaget är rimliga. Detta gör det väl lämpat för höggenomströmnings- och autonoma laboratorier, där robotar kan syntetisera nya föreningar, mäta deras XRD-mönster och låta XCCP föreslå troliga strukturer i realtid, vilket snabbar upp vägen från rådata till nya material.
Citering: Xu, C., Su, T., Xiong, J. et al. KAN-enhanced contrastive learning: the accelerator of crystal structure identification from XRD patterns. npj Comput Mater 12, 144 (2026). https://doi.org/10.1038/s41524-026-02015-y
Nyckelord: pulverröntgendiffraktion, identifiering av kristallstruktur, kontrastiv inlärning, materialinformatik, Kolmogorov–Arnold-nätverk