Clear Sky Science · de
KAN-verbessertes kontrastives Lernen: der Beschleuniger der Kristallstrukturidentifikation aus XRD‑Mustern
Warum schnellere Kristallkartierung wichtig ist
Neue Materialien für Batterien, Elektronik und saubere Energie werden oft ein Kristall nach dem anderen entdeckt. Die interne Anordnung der Atome bestimmt das Verhalten jedes Kristalls, und Wissenschaftler lesen diese Anordnung in der Regel aus Pulver-Röntgendiffraktions (XRD)-Mustern ab — spitzenreiche Grafiken, die entstehen, wenn Röntgenstrahlen von einer Probe gestreut werden. Heute ist die Umwandlung dieser Muster in eine konkrete Struktur ein langsamer, expertenintensiver Prozess. Diese Arbeit stellt ein maschinelles Lernsystem vor, das ein XRD‑Muster rasch mit wahrscheinlichen Kristallstrukturen abgleichen kann, wodurch diese Detektivarbeit schneller, zuverlässiger und leichter in automatisierte Labore integrierbar wird.
Von spitzenreichen Mustern zu atomaren Blaupausen
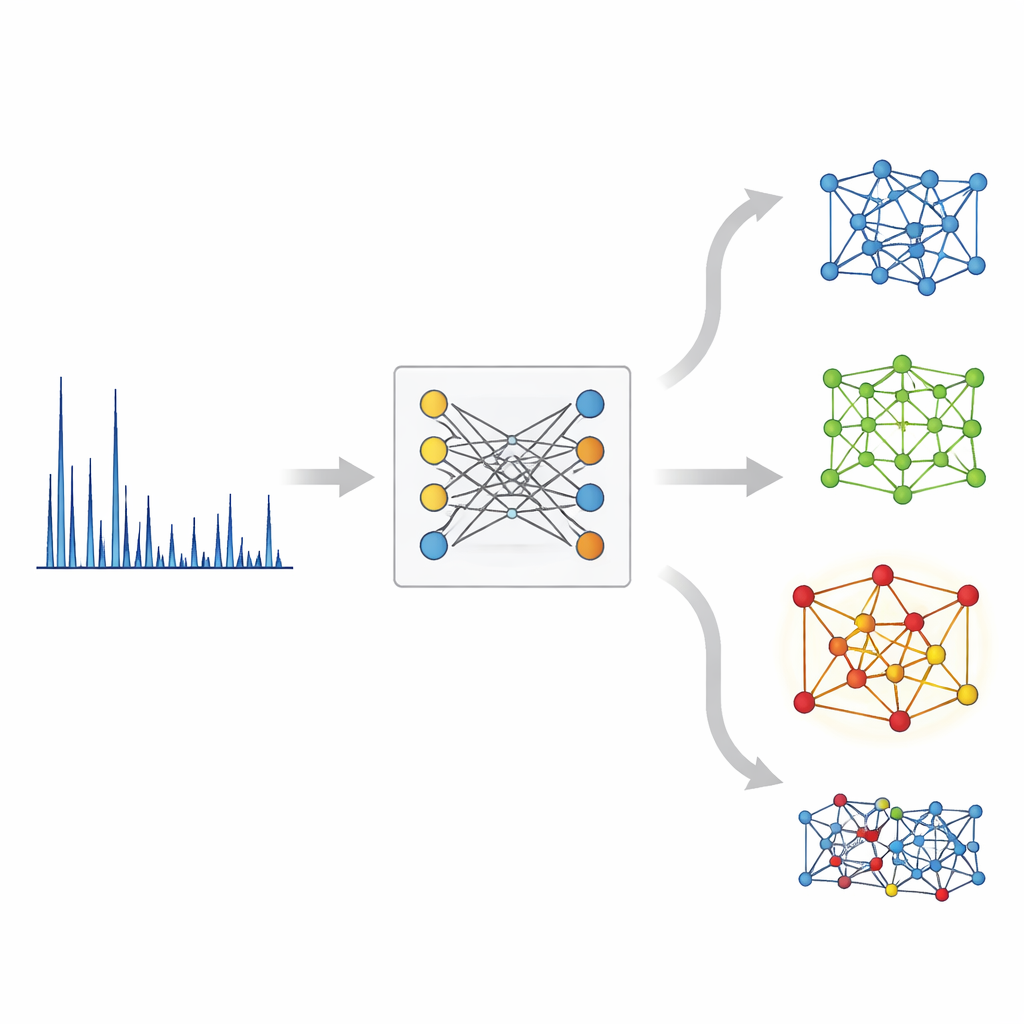
In der konventionellen Praxis inspiziert ein XRD-Spezialist die Peaks eines Musters, nutzt physikalische Formeln, um mögliche Atomabstände herzuleiten, und vergleicht dann iterativ Kandidatenstrukturen mit den Daten. Dieser Prozess gerät an seine Grenzen, wenn Peaks überlappen oder viele ähnliche Möglichkeiten existieren, und er skaliert schlecht für moderne Hochdurchsatz-Experimente, die tausende Muster pro Tag erzeugen können. Frühere maschinelle Lernwerkzeuge behandelten XRD meist wie ein Kennzeichnungsproblem — sie sagten eine Symmetrieklasse oder Raumgruppe aus einem Muster voraus — statt direkt die Struktur selbst zu identifizieren. Der neue Ansatz, XRD‑Crystal Contrastive Pretraining (XCCP) genannt, formuliert die Aufgabe als Retrieval: Gegeben ein Muster, finde den am besten passenden Kristall in einer großen Datenbank.
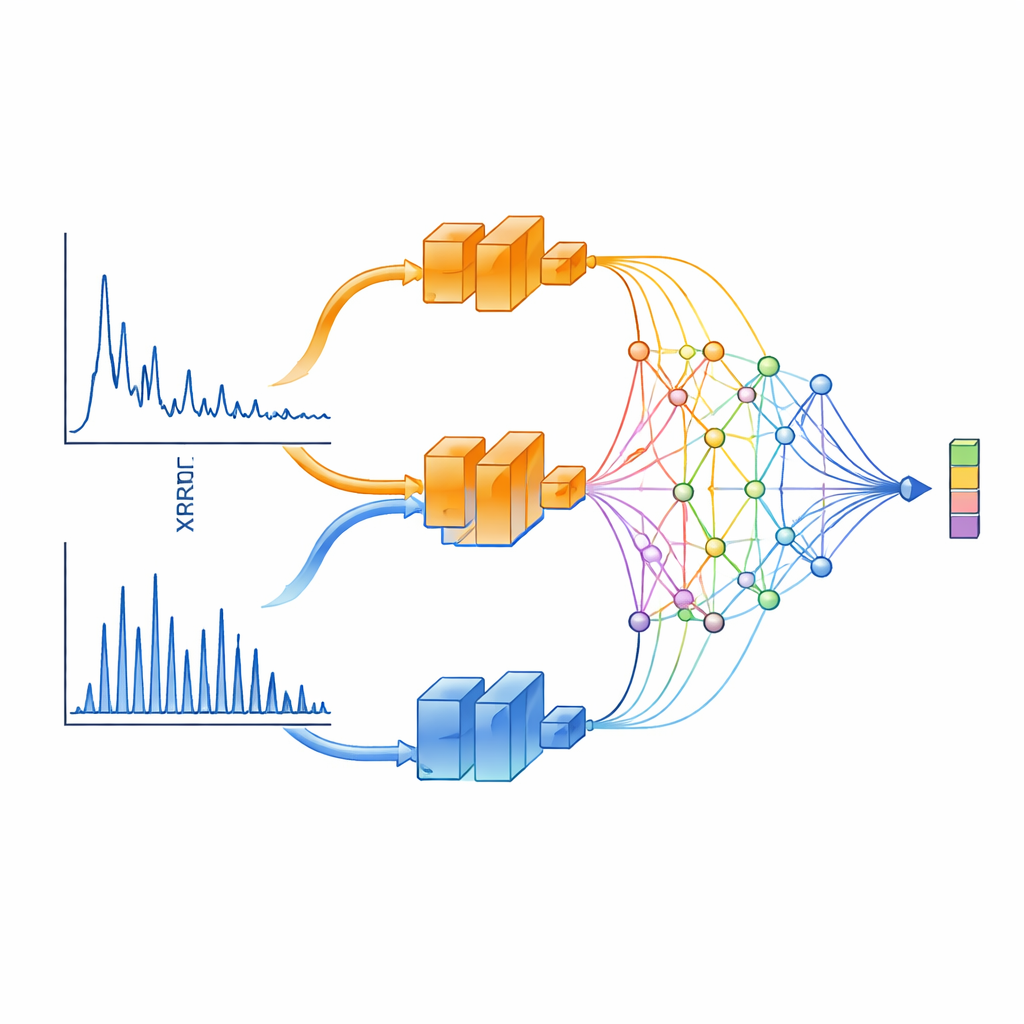
Ein zweiaugiger Blick auf Röntgenmuster
XCCP lernt, XRD-Daten physikalisch informiert zu „sehen“. Anstatt das gesamte Muster in ein einzelnes neuronales Netz zu speisen, teilt die Methode es in zwei Bereiche auf. Ein Zweig konzentriert sich auf kleine Winkel, die langreichweitige Merkmale wie geschichtete Abstände und Supergitter erfassen. Der andere fokussiert auf große Winkel, wo die Peaks dicht liegen und stark von der Kristallsymmetrie bestimmt werden. Jeder Zweig wird von einem tiefen Netzwerk verarbeitet und anschließend durch ein spezielles Projektionsmodul auf Basis von Kolmogorov–Arnold-Netzwerken (KANs) kombiniert. Dieses Modul ist besonders gut darin, Aufmerksamkeit auf schmale Bereiche des Musters zu lenken — genau dorthin, wo scharfe Beugungspeaks die meisten strukturellen Informationen tragen.
Muster und Strukturen in der Mitte zusammenführen
Auf der Kristallseite verwendet XCCP ein graphbasiertes Netzwerk, das Atome als Knoten und ihre Bindungen als Verbindungen darstellt. Während des Trainings sieht das System viele gepaarte Beispiele: ein XRD‑Muster und seine bekannte Kristallstruktur. Es lernt einen gemeinsamen numerischen Raum, in dem jedes Muster nah an seiner eigenen Struktur und weit von nicht passenden liegt. Wenn ein neues Muster eingeht, bettet das Modell es in diesem Raum ein, vergleicht es mit den Einbettungen aller Datenbankstrukturen und liefert eine gerankte Auswahlliste. Ohne Kenntnis der vorhandenen Elemente steht die korrekte Struktur fast zur Hälfte der Fälle an erster Stelle und erscheint für die überwiegende Mehrheit der Fälle in den Top fünf. Wenn der Nutzer zusätzlich die chemische Zusammensetzung angibt — Informationen, die in realen Experimenten üblicherweise verfügbar sind — ist der Top‑1‑Treffer fast 90 % der Zeit korrekt.
Sehen, was die Maschine sieht
Die Autoren untersuchen, ob ihr System sich auf echte Physik oder auf zufällige Datenbesonderheiten stützt. Durch Maskieren von Musternteilen und den Einsatz von Attributionswerkzeugen zeigen sie, dass der KAN-Kopf seine Entscheidungen hauptsächlich auf starke, gut definierte Beugungspeaks stützt und nicht auf breite Hintergrundvariationen oder Rauschen. Der hinzugefügte Niedrigwinkelzweig verbessert konsistent die Leistung, insbesondere bei niedrigsymmetrischen Kristallen und Mustern, in denen hochwinkelige Merkmale mehrdeutig sind. Das Modell erweist sich zudem als robust gegenüber gängigen experimentellen Unvollkommenheiten wie Peakverbreiterung und kleinen Verschiebungen entlang der Winkelachse und überträgt sich recht gut auf reale experimentelle Datensätze. Wichtig ist, dass die erzeugten Ähnlichkeitswerte auch als Vertrauensmaß dienen — sie fallen deutlich ab, wenn die wahre Struktur nicht in der Datenbank vorhanden ist, eine wesentliche Eigenschaft für einen sicheren Einsatz in der Praxis.
Auf dem Weg zu intelligenterer, selbstfahrender Materialentdeckung
Für einen Nicht‑Spezialisten lautet die Hauptbotschaft: XCCP wandelt die XRD‑Analyse von einer Handwerkskunst in eine schnelle, datengetriebene Suche. Indem Beugungsmuster und Kandidatenkristalle in einem gemeinsamen Raum ausgerichtet werden und ein physikbewusstes Netzwerkdesign verwendet wird, kann das System schnell eine kurze Liste realistischer atomarer Blaupausen mit interpretierbarer Vertrauensangabe vorschlagen. Es ersetzt nicht das fachliche Urteil oder detaillierte Verfeinerungen, beschleunigt jedoch erheblich den ersten, schwierigsten Schritt — herauszufinden, welche Strukturen überhaupt plausibel sind. Das macht es gut geeignet für Hochdurchsatz- und autonome Labore, in denen Roboter neue Verbindungen synthetisieren, deren XRD‑Muster messen und XCCP in Echtzeit wahrscheinliche Strukturen vorschlagen lässt, wodurch der Weg von Rohdaten zu neuen Materialien verkürzt wird.
Zitation: Xu, C., Su, T., Xiong, J. et al. KAN-enhanced contrastive learning: the accelerator of crystal structure identification from XRD patterns. npj Comput Mater 12, 144 (2026). https://doi.org/10.1038/s41524-026-02015-y
Schlüsselwörter: Pulver-Röntgendiffraktion, Kristallstrukturidentifikation, kontrastives Lernen, Materialinformatik, Kolmogorov–Arnold-Netzwerke