Clear Sky Science · pt
KAN-aprimorado contraste de aprendizado: o acelerador da identificação de estruturas cristalinas a partir de padrões de DRX
Por que o mapeamento cristalino mais rápido importa
Novos materiais para baterias, eletrônica e energia limpa são frequentemente descobertos um cristal de cada vez. O arranjo atômico interno de cada cristal determina seu comportamento, e os cientistas geralmente leem esse arranjo a partir de padrões de difração de raios X em pó (DRX) — gráficos com picos gerados quando raios X espalham-se a partir de uma amostra. Hoje, transformar esses padrões em uma estrutura concreta é um trabalho lento e dependente de especialistas. Este artigo apresenta um sistema de aprendizado de máquina que pode rapidamente emparelhar um padrão de DRX com estruturas cristalinas prováveis, tornando esse trabalho de detetive mais rápido, mais confiável e mais fácil de integrar em laboratórios automatizados.
De padrões picudos a plantas atômicas
Na prática convencional, um especialista em DRX inspeciona os picos de um padrão, usa fórmulas físicas para inferir possíveis espaçamentos atômicos e então compara iterativamente estruturas candidatas com os dados. Esse processo enfrenta dificuldades quando os picos se sobrepõem ou quando há muitas possibilidades semelhantes, e não escala bem para experimentos modernos de alto rendimento que podem produzir milhares de padrões por dia. Ferramentas de aprendizado de máquina anteriores trataram a DRX em grande parte como um problema de rotulagem — prevendo uma classe de simetria ou grupo espacial a partir de um padrão — em vez de identificar diretamente a estrutura em si. A nova abordagem, chamada XRD‑Crystal Contrastive Pretraining (XCCP), reformula a tarefa como recuperação: dado um padrão, encontre o cristal mais compatível em um grande banco de dados.
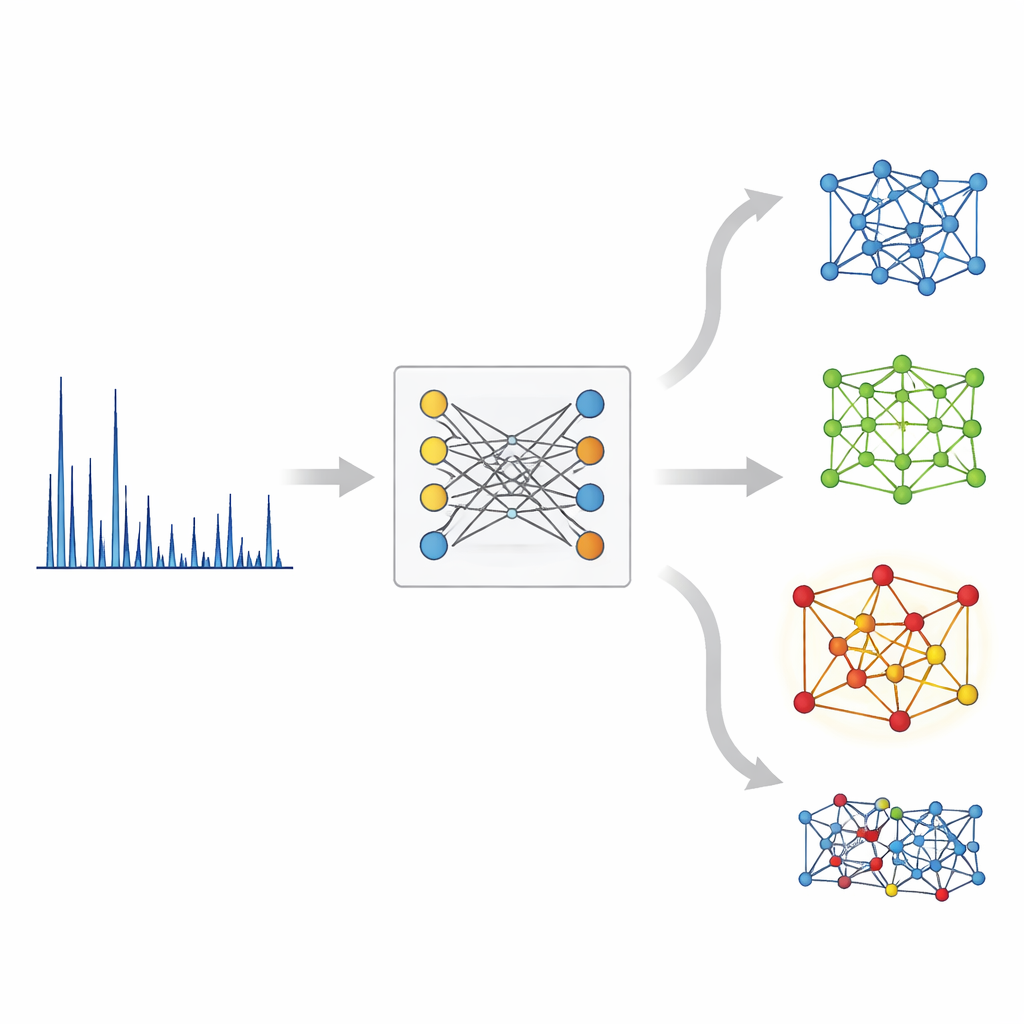
Uma visão binocular dos padrões de raios X
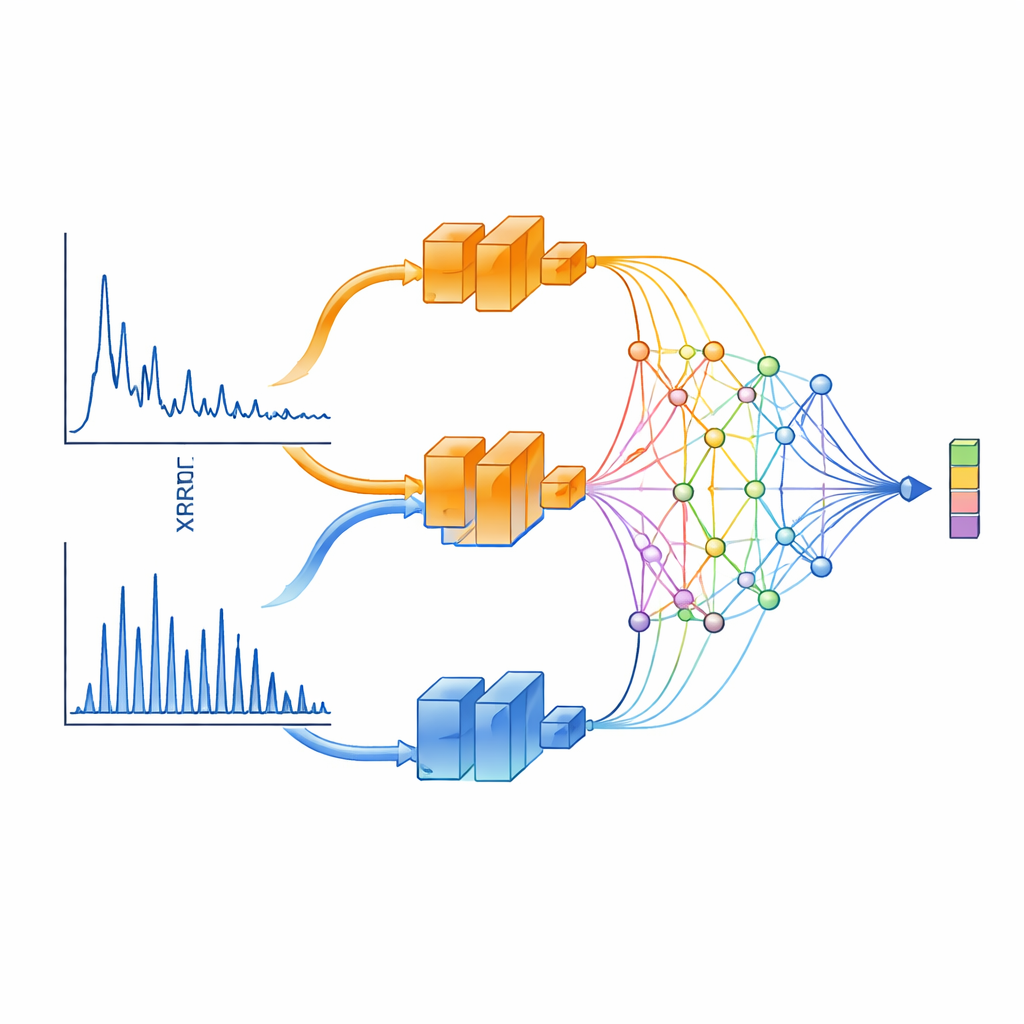
O XCCP aprende a “ver” dados de DRX de maneira informada pela física. Em vez de alimentar o padrão inteiro em uma única rede neural, o método o divide em dois intervalos. Um ramo foca em ângulos pequenos, que capturam características de longa distância como espaçamentos em camadas e super-redes. O outro concentra-se em ângulos amplos, onde os picos são densos e fortemente governados pela simetria cristalina. Cada ramo é processado por uma rede profunda e então combinado por um módulo de projeção especial baseado em Kolmogorov–Arnold Networks (KANs). Esse módulo se destaca em focar atenção em regiões estreitas do padrão — precisamente onde picos nítidos de difração carregam a maior parte da informação estrutural.
Deixando padrões e estruturas se encontrarem no meio
No lado cristalino, o XCCP usa uma rede baseada em grafos que representa átomos como nós e suas ligações como conexões. Durante o treinamento, o sistema vê muitos pares correspondentes: um padrão de DRX e sua estrutura cristalina conhecida. Ele aprende um espaço numérico compartilhado onde cada padrão fica próximo de sua própria estrutura e distante de pares não correspondentes. Quando um novo padrão chega, o modelo o incorpora nesse espaço, compara-o com embeddings de todas as estruturas do banco de dados e retorna uma lista ranqueada. Sem qualquer conhecimento dos elementos presentes, a estrutura correta é classificada em primeiro lugar quase na metade das vezes e aparece entre os cinco primeiros na grande maioria dos casos. Quando o usuário também fornece a composição química — informação comumente disponível em experimentos reais — a correspondência top‑1 está correta em quase 90% das vezes.
Vendo o que a máquina vê
Os autores investigam se seu sistema se apoia em física real ou em peculiaridades acidentais dos dados. Ao mascarar partes do padrão e usar ferramentas de atribuição, mostram que a cabeça KAN baseia suas decisões principalmente em picos de difração fortes e bem definidos, em vez de variações amplas de fundo ou ruído. O ramo de baixo ângulo adicional melhora consistentemente o desempenho, especialmente para cristais de baixa simetria e padrões onde características de alto ângulo são ambíguas. O modelo também se mostra robusto a imperfeições experimentais comuns, como alargamento de picos e pequenos deslocamentos ao longo do eixo de ângulo, e transfere-se razoavelmente bem para conjuntos de dados experimentais reais. Importante, as pontuações de similaridade que produz funcionam também como medidas de confiança, caindo de forma marcada quando a estrutura verdadeira está ausente do banco de dados — uma propriedade essencial para uso seguro no mundo real.
Rumo a uma descoberta de materiais mais inteligente e autônoma
Para um não especialista, a mensagem principal é que o XCCP transforma a análise de DRX de um ofício em uma busca rápida orientada por dados. Ao alinhar padrões de difração e cristais candidatos em um espaço compartilhado, e ao usar um projeto de rede sensível à física, o sistema pode propor rapidamente uma lista curta de plantas atômicas realistas com confiança interpretável. Ele não substitui o julgamento de especialistas nem refinamentos detalhados, mas acelera consideravelmente o primeiro e mais difícil passo — descobrir quais estruturas são sequer plausíveis. Isso o torna bem adequado para laboratórios de alto rendimento e autônomos, onde robôs podem sintetizar novos compostos, medir seus padrões de DRX e permitir que o XCCP sugira estruturas prováveis em tempo real, acelerando o caminho dos dados brutos a novos materiais.
Citação: Xu, C., Su, T., Xiong, J. et al. KAN-enhanced contrastive learning: the accelerator of crystal structure identification from XRD patterns. npj Comput Mater 12, 144 (2026). https://doi.org/10.1038/s41524-026-02015-y
Palavras-chave: difração de raios X em pó, identificação de estrutura cristalina, aprendizado contrastivo, informática de materiais, redes de Kolmogorov–Arnold