Clear Sky Science · it
Apprendimento contrastivo potenziato da KAN: l’acceleratore per l’identificazione della struttura cristallina da spettri XRD
Perché mappare i cristalli più velocemente è importante
I nuovi materiali per batterie, elettronica ed energie pulite vengono spesso scoperti un cristallo alla volta. L’assetto atomico interno di ciascun cristallo determina il suo comportamento, e gli scienziati normalmente ricavano questo assetto da spettri di diffrazione a raggi X (XRD) su polveri—grafici a picchi prodotti dalla diffusione dei raggi X su un campione. Oggi trasformare quei pattern in una struttura concreta è un lavoro lento e che richiede esperti. Questo articolo presenta un sistema di machine learning in grado di abbinare rapidamente uno spettro XRD alle strutture cristalline più probabili, rendendo questo lavoro investigativo più veloce, più affidabile e più facile da integrare in laboratori automatizzati.
Dai pattern acuminati ai progetti atomici
Nella pratica convenzionale, uno specialista XRD ispeziona i picchi di un pattern, usa formule fisiche per dedurre possibili distanze atomiche e poi confronta iterativamente strutture candidate con i dati. Questo processo è problematico quando i picchi si sovrappongono o quando esistono molte possibilità simili, e non scala bene agli esperimenti moderni ad alta produttività che possono produrre migliaia di pattern al giorno. Gli strumenti di machine learning passati hanno in gran parte trattato l’XRD come un problema di etichettatura—predire una classe di simmetria o un gruppo spaziale da un pattern—invece di identificare direttamente la struttura. Il nuovo approccio, chiamato XRD‑Crystal Contrastive Pretraining (XCCP), riformula il compito come retrieval: dato un pattern, trova il cristallo più compatibile in un ampio database.
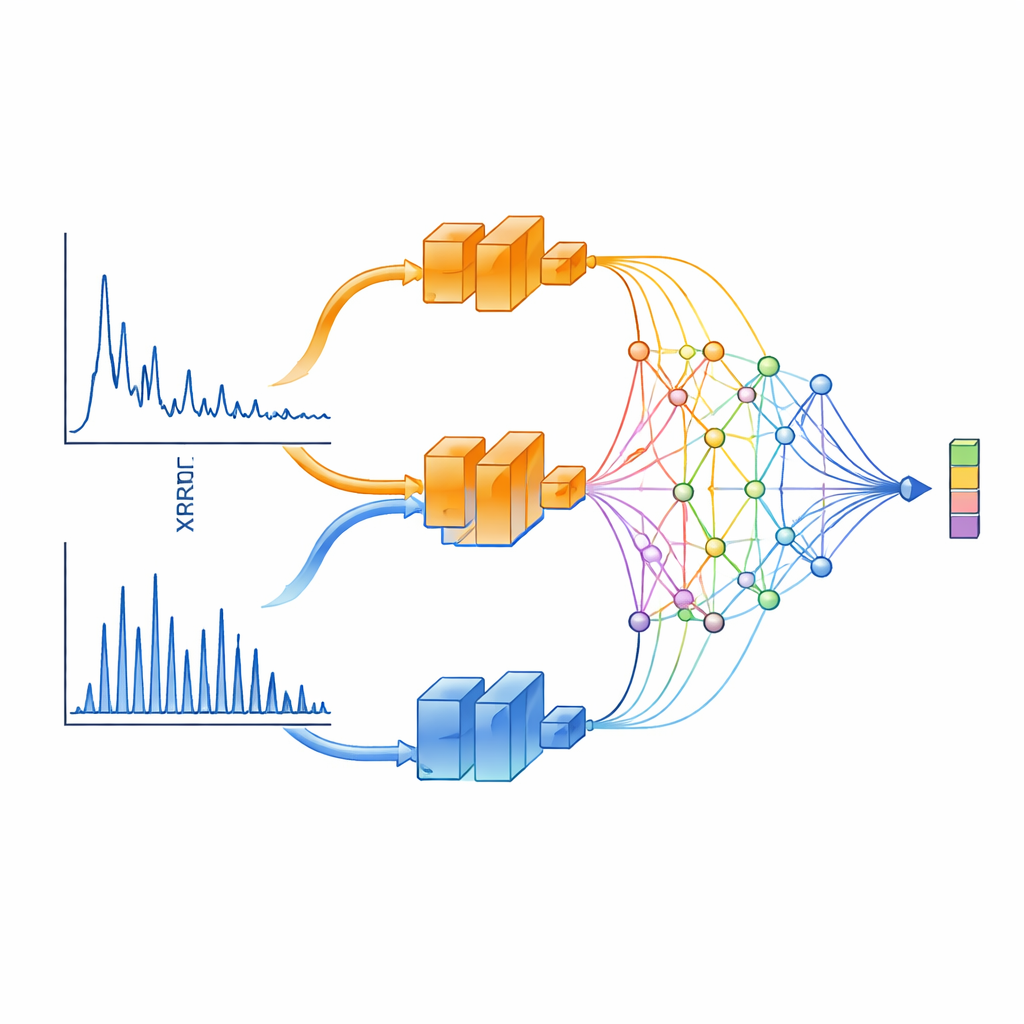
Una visione a due sguardi degli spettri a raggi X
XCCP impara a “vedere” i dati XRD in modo informato dalla fisica. Invece di alimentare l’intero pattern in una singola rete neurale, il metodo lo suddivide in due intervalli. Un ramo si concentra sugli angoli piccoli, che catturano caratteristiche a lunga distanza come spaziature a strati e superreticoli. L’altro si concentra sugli angoli ampi, dove i picchi sono densi e fortemente determinati dalla simmetria cristallina. Ogni ramo è processato da una rete profonda e poi combinato tramite un modulo di proiezione speciale basato su Kolmogorov–Arnold Networks (KAN). Questo modulo eccelle nell’attenzionare regioni strette del pattern—proprio dove i picchi di diffrazione netti contengono le informazioni strutturali più significative.
Far incontrare pattern e strutture a metà strada
Sul fronte delle strutture, XCCP utilizza una rete basata su grafi che rappresenta gli atomi come nodi e i loro legami come connessioni. Durante l’addestramento, il sistema vede molte coppie abbinate: uno spettro XRD e la sua struttura cristallina nota. Impara uno spazio numerico condiviso in cui ogni pattern è vicino alla propria struttura e lontano da quelle non corrispondenti. Quando arriva un nuovo pattern, il modello lo incorpora in questo spazio, lo confronta con gli embedding di tutte le strutture nel database e restituisce una lista classificata. Senza alcuna informazione sugli elementi chimici presenti, la struttura corretta è al primo posto quasi la metà delle volte e compare nelle prime cinque per la stragrande maggioranza dei casi. Quando l’utente fornisce anche la composizione chimica—informazione comunemente disponibile negli esperimenti reali—la corrispondenza al primo posto è corretta quasi il 90% delle volte.
Vedere ciò che vede la macchina
Gli autori indagano se il sistema si basi sulla fisica reale o su peculiarità accidentali dei dati. Mascherando parti del pattern e usando strumenti di attribuzione, mostrano che la testa KAN fonda le sue decisioni principalmente su picchi di diffrazione forti e ben definiti piuttosto che su variazioni di fondo ampie o rumore. Il ramo a basso angolo aggiunto migliora costantemente le prestazioni, specialmente per cristalli a bassa simmetria e per pattern in cui le caratteristiche ad alto angolo sono ambigue. Il modello si dimostra inoltre robusto a imperfezioni sperimentali comuni come l’allargamento dei picchi e piccoli spostamenti lungo l’asse angolare, e trasferisce ragionevolmente bene alle serie di dati sperimentali reali. È importante che i punteggi di similarità prodotti fungano anche da misure di confidenza, scendendo nettamente quando la vera struttura è assente dal database—una proprietà essenziale per un uso sicuro nel mondo reale.
Verso una scoperta dei materiali più intelligente e autonoma
Per un non specialista, il messaggio principale è che XCCP trasforma l’analisi XRD da un mestiere in una ricerca rapida e guidata dai dati. Allineando pattern di diffrazione e cristalli candidati in uno spazio condiviso e adottando un design di rete consapevole della fisica, il sistema può rapidamente proporre una breve lista di progetti atomici realistici con confidenze interpretabili. Non sostituisce il giudizio dell’esperto o il raffinamento dettagliato, ma accelera notevolmente il primo e più difficile passo—capire quali strutture sono anche solo plausibili. Questo lo rende particolarmente adatto per laboratori ad alta produttività e autonomi, dove robot possono sintetizzare nuovi composti, misurarne gli spettri XRD e lasciare che XCCP suggerisca in tempo reale le strutture più probabili, accelerando il percorso dai dati grezzi a nuovi materiali.
Citazione: Xu, C., Su, T., Xiong, J. et al. KAN-enhanced contrastive learning: the accelerator of crystal structure identification from XRD patterns. npj Comput Mater 12, 144 (2026). https://doi.org/10.1038/s41524-026-02015-y
Parole chiave: diffrazione di raggi X da polveri, identificazione della struttura cristallina, apprendimento contrastivo, informatica dei materiali, reti di Kolmogorov–Arnold