Clear Sky Science · en
KAN-enhanced contrastive learning: the accelerator of crystal structure identification from XRD patterns
Why Faster Crystal Mapping Matters
New materials for batteries, electronics, and clean energy are often discovered one crystal at a time. Each crystal’s internal atomic arrangement determines how it behaves, and scientists usually read this arrangement from powder X‑ray diffraction (XRD) patterns—spiky graphs produced when X‑rays scatter from a sample. Today, turning those patterns into a concrete structure is slow, expert-heavy work. This paper introduces a machine-learning system that can rapidly match an XRD pattern to likely crystal structures, making this detective work faster, more reliable, and easier to plug into automated labs.
From Spiky Patterns to Atomic Blueprints
In conventional practice, an XRD specialist inspects a pattern’s peaks, uses physics formulas to infer possible atomic spacings, and then iteratively compares candidate structures against the data. This process struggles when peaks overlap or when there are many similar possibilities, and it does not scale well to modern high-throughput experiments that can produce thousands of patterns per day. Past machine-learning tools have mostly treated XRD like a labeling problem—predicting a symmetry class or space group from a pattern—rather than directly identifying the structure itself. The new approach, called XRD‑Crystal Contrastive Pretraining (XCCP), reframes the task as retrieval: given a pattern, find the most compatible crystal in a large database.
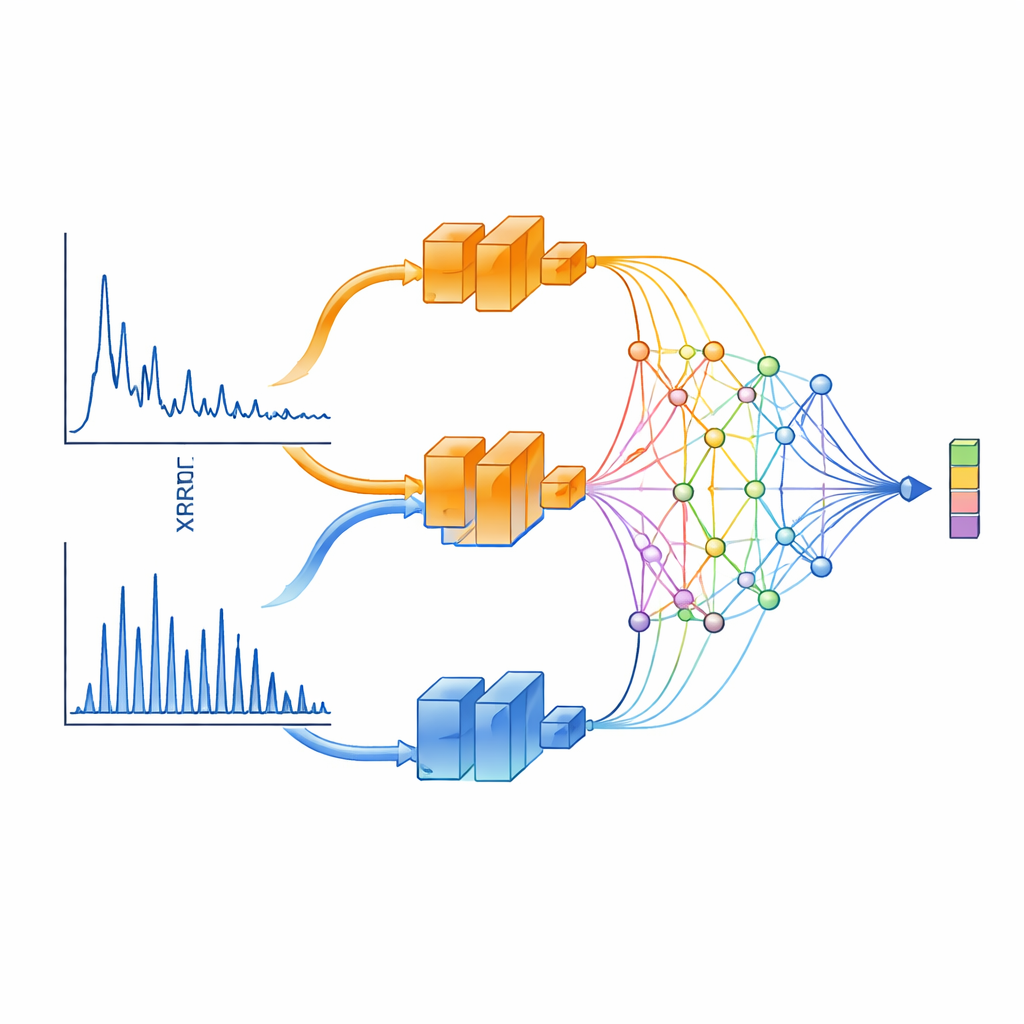
A Two-Eyed View of X‑Ray Patterns
XCCP learns to “see” XRD data in a physically informed way. Instead of feeding the entire pattern into a single neural network, the method splits it into two ranges. One branch focuses on small angles, which capture long-distance features like layered spacings and superlattices. The other concentrates on wide angles, where peaks are dense and strongly governed by crystal symmetry. Each branch is processed by a deep network and then combined by a special projection module based on Kolmogorov–Arnold Networks (KANs). This module excels at focusing attention on narrow regions of the pattern—precisely where sharp diffraction peaks carry the most structural information.
Letting Patterns and Structures Meet in the Middle
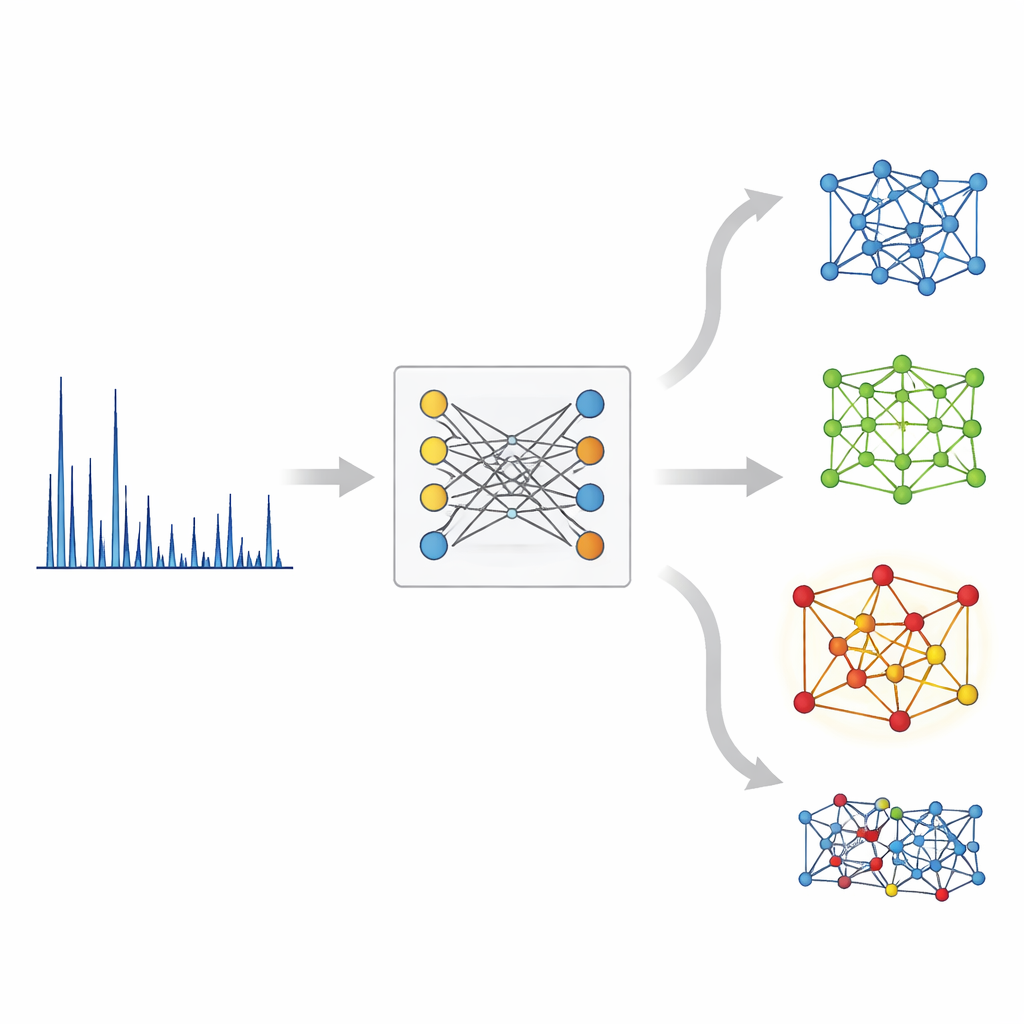
On the crystal side, XCCP uses a graph-based network that represents atoms as nodes and their bonds as connections. During training, the system sees many matched pairs: an XRD pattern and its known crystal structure. It learns a shared numerical space where each pattern sits close to its own structure and far from mismatched ones. When a new pattern arrives, the model embeds it into this space, compares it with embeddings of all database structures, and returns a ranked shortlist. Without any knowledge of which elements are present, the correct structure is ranked first nearly half the time and appears in the top five for the vast majority of cases. When the user also supplies the chemical composition—information commonly available in real experiments—the top‑1 match is correct almost 90% of the time.
Seeing What the Machine Sees
The authors probe whether their system is relying on real physics or on accidental quirks of the data. By masking parts of the pattern and using attribution tools, they show that the KAN head bases its decisions mainly on strong, well-defined diffraction peaks rather than on broad background variations or noise. The added low-angle branch consistently improves performance, especially for low-symmetry crystals and patterns where high-angle features are ambiguous. The model also proves robust to common experimental imperfections such as peak broadening and small shifts along the angle axis, and it transfers reasonably well to real experimental datasets. Importantly, the similarity scores it produces double as confidence measures, dropping markedly when the true structure is absent from the database—an essential property for safe, real-world use.
Toward Smarter, Self-Driving Materials Discovery
For a non-specialist, the main message is that XCCP turns XRD analysis from a craft into a fast, data-driven search. By aligning diffraction patterns and candidate crystals in a shared space, and by using physics-aware network design, the system can rapidly propose a short list of realistic atomic blueprints with interpretable confidence. It does not replace expert judgment or detailed refinement, but it greatly accelerates the first, hardest step—figuring out which structures are even plausible. This makes it well-suited for high-throughput and autonomous laboratories, where robots can synthesize new compounds, measure their XRD patterns, and let XCCP suggest likely structures in real time, speeding up the path from raw data to new materials.
Citation: Xu, C., Su, T., Xiong, J. et al. KAN-enhanced contrastive learning: the accelerator of crystal structure identification from XRD patterns. npj Comput Mater 12, 144 (2026). https://doi.org/10.1038/s41524-026-02015-y
Keywords: powder X-ray diffraction, crystal structure identification, contrastive learning, materials informatics, Kolmogorov–Arnold networks