Clear Sky Science · pl
KAN-wzmocnione uczenie kontrastowe: akcelerator identyfikacji struktury krystalicznej na podstawie wzorców dyfrakcji rentgenowskiej
Dlaczego szybsze mapowanie kryształów ma znaczenie
Nowe materiały do baterii, elektroniki i technologii czystej energii często odkrywane są po jednym kryształu naraz. Wewnętrzne uporządkowanie atomów w każdym krysztale determinuje jego właściwości, a naukowcy zwykle odczytują to uporządkowanie z proszkowych wzorców dyfrakcji rentgenowskiej (XRD) — kolczastych wykresów powstających, gdy promienie rentgenowskie rozpraszają się na próbce. Dziś przekształcenie tych wzorców w konkretną strukturę jest procesem powolnym i wymagającym specjalistycznej wiedzy. W artykule przedstawiono system uczenia maszynowego, który szybko dopasowuje wzorzec XRD do prawdopodobnych struktur krystalicznych, przyspieszając tę detektywistyczną pracę, zwiększając jej niezawodność i ułatwiając integrację z zautomatyzowanymi laboratoriami.
Z kolczastych wykresów do atomowych planów
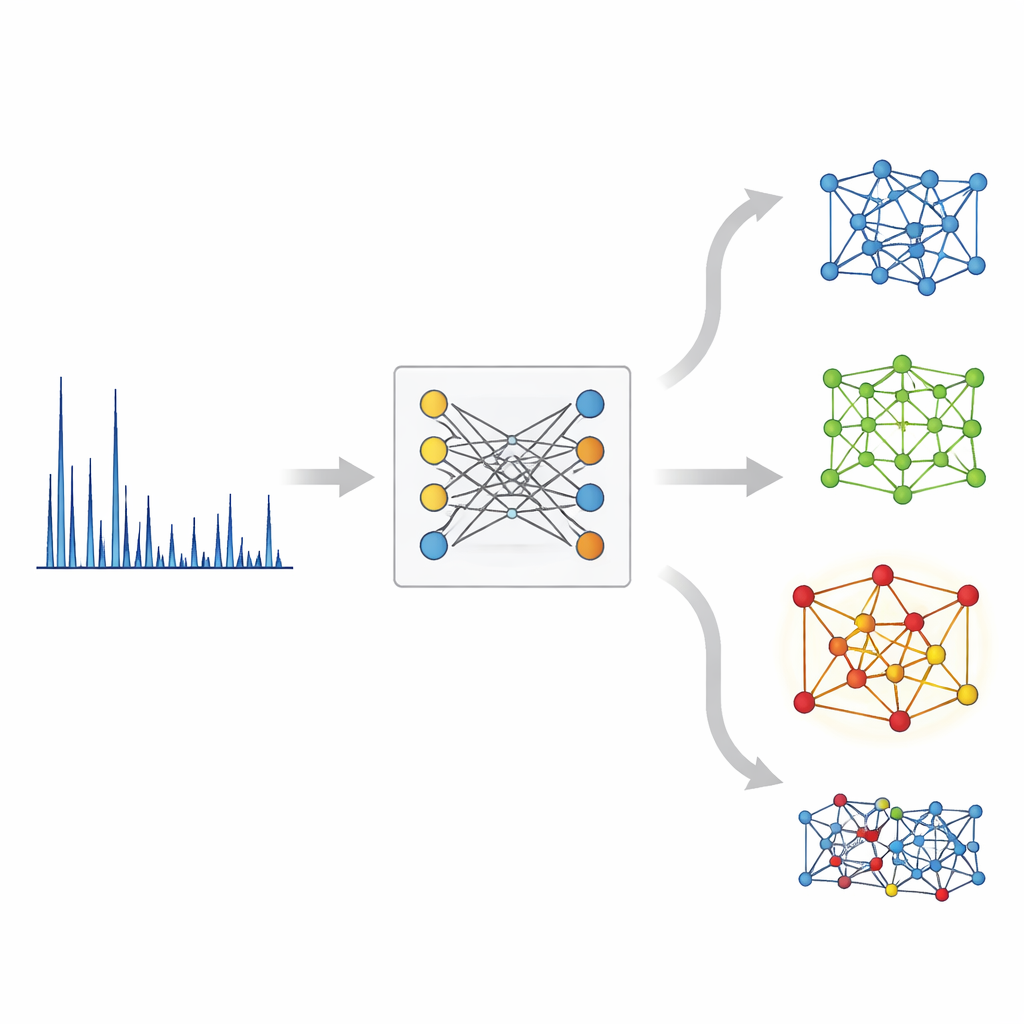
W konwencjonalnej praktyce specjalista XRD analizuje piki wzorca, używa równań fizycznych do wywnioskowania możliwych odległości międzyatomowych, a następnie iteracyjnie porównuje kandydatów struktur z danymi. Proces ten napotyka trudności, gdy piki nakładają się lub gdy istnieje wiele podobnych możliwości, i słabo skaluje się do nowoczesnych eksperymentów o dużej przepustowości, które mogą generować tysiące wzorców dziennie. Dotychczasowe narzędzia uczenia maszynowego traktowały XRD głównie jako problem etykietowania — przewidywanie klasy symetrii lub grupy przestrzennej na podstawie wzorca — zamiast bezpośredniej identyfikacji struktury. Nowe podejście, nazwane XRD‑Crystal Contrastive Pretraining (XCCP), przedefiniowuje zadanie jako wyszukiwanie: mając wzorzec, znajdź najbardziej kompatybilny kryształ w dużej bazie danych.
Dwuwymiarowe spojrzenie na wzorce rentgenowskie
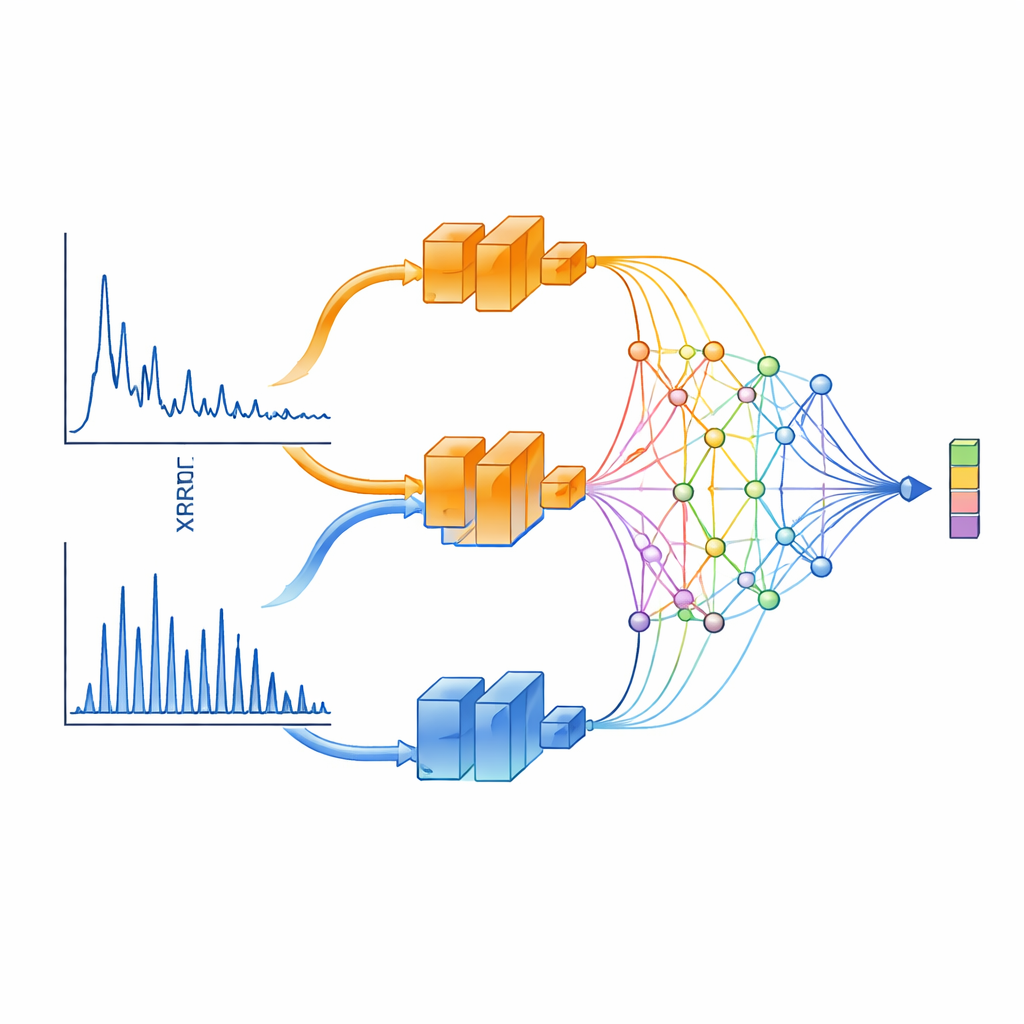
XCCP uczy się „widzieć” dane XRD w sposób uwzględniający fizykę. Zamiast podawać cały wzorzec do jednej sieci neuronowej, metoda dzieli go na dwa zakresy. Jedna gałąź koncentruje się na małych kątach, które wychwytują cechy na długich dystansach, takie jak odległości warstwowe i nadstruktury. Druga skupia się na szerokich kątach, gdzie piki są gęste i w dużym stopniu determinowane przez symetrię kryształu. Każda z gałęzi jest przetwarzana przez głęboką sieć, a następnie łączona przez specjalny moduł projekcji oparty na Sieciach Kołmogorowa–Arnolda (KAN). Ten moduł doskonale skupia uwagę na wąskich obszarach wzorca — dokładnie tam, gdzie ostre piki dyfrakcyjne niosą najwięcej informacji strukturalnej.
Pozwalając wzorcom i strukturom spotkać się w połowie drogi
Po stronie struktury XCCP używa sieci grafowej, która reprezentuje atomy jako węzły, a ich wiązania jako połączenia. Podczas treningu system widzi wiele sparowanych par: wzorzec XRD i jego znana struktura krystaliczna. Uczy się wspólnej przestrzeni numerycznej, w której każdy wzorzec znajduje się blisko swojej struktury i daleko od dopasowań nieprawidłowych. Gdy pojawia się nowy wzorzec, model osadza go w tej przestrzeni, porównuje jego osadzenia ze wszystkimi osadzeniami struktur z bazy danych i zwraca uporządkowaną listę. Bez wiedzy o obecności konkretnych pierwiastków poprawna struktura znajduje się na pierwszym miejscu niemal w połowie przypadków, a w większości przypadków pojawia się w top 5. Gdy użytkownik poda też skład chemiczny — informację często dostępną w rzeczywistych eksperymentach — trafność top‑1 wynosi prawie 90%.
Widzieć to, co widzi maszyna
Autorzy badają, czy ich system opiera się na rzeczywistej fizyce, czy na przypadkowych cechach danych. Poprzez maskowanie fragmentów wzorca i użycie narzędzi atrybucji wykazują, że głowa KAN podejmuje decyzje głównie na podstawie silnych, dobrze zdefiniowanych pików dyfrakcyjnych, a nie na podstawie szerokich zmian tła czy szumu. Dodatkowa gałąź niskokątowa konsekwentnie poprawia wydajność, szczególnie dla kryształów o niskiej symetrii i wzorców, w których cechy wysokokątowe są niejednoznaczne. Model okazuje się także odporny na typowe niedoskonałości eksperymentalne, takie jak rozszerzanie się pików i niewielkie przesunięcia w osi kąta, oraz przenosi się w miarę dobrze na rzeczywiste zbiory danych eksperymentalnych. Co ważne, wygenerowane przez model miary podobieństwa służą jednocześnie jako miary pewności, które znacząco spadają, gdy prawdziwej struktury brakuje w bazie danych — istotna cecha dla bezpiecznego stosowania w praktyce.
W kierunku mądrzejszego, samojezdnego odkrywania materiałów
Dla osoby niebędącej specjalistą główny wniosek jest taki, że XCCP przekształca analizę XRD z rzemiosła w szybkie, napędzane danymi wyszukiwanie. Poprzez wyrównanie wzorców dyfrakcyjnych i kandydatów na struktury w wspólnej przestrzeni oraz zastosowanie projektowania sieci uwzględniającego fizykę, system może szybko zaproponować krótką listę realistycznych atomowych planów z interpretowalnymi miarami pewności. Nie zastępuje to oceny eksperckiej ani szczegółowej refinacji, ale znacznie przyspiesza pierwszy, najtrudniejszy krok — ustalenie, które struktury w ogóle są prawdopodobne. Czyni to rozwiązanie szczególnie przydatnym w laboratoriach o dużej przepustowości i autonomicznych, gdzie roboty mogą syntetyzować nowe związki, mierzyć ich wzorce XRD, a XCCP sugerować prawdopodobne struktury w czasie rzeczywistym, przyspieszając drogę od surowych danych do nowych materiałów.
Cytowanie: Xu, C., Su, T., Xiong, J. et al. KAN-enhanced contrastive learning: the accelerator of crystal structure identification from XRD patterns. npj Comput Mater 12, 144 (2026). https://doi.org/10.1038/s41524-026-02015-y
Słowa kluczowe: proszkowa dyfrakcja rentgenowska, identyfikacja struktury krystalicznej, uczenie kontrastowe, informatyka materiałowa, sieci Kołmogorowa–Arnolda