Clear Sky Science · sv
Stereoselektiv vicinal C(sp³)–C-bindningsbildning via metallafotoredox 1,2-difunktionalisering av interna alkener
Bygga komplexa molekyler med ljus
Kemister behöver ofta sammanfoga små kolfragment till intrikata tredimensionella former för läkemedel och material. Att göra detta snabbt, rent och kontrollerat är förvånansvärt svårt, särskilt när två nya kol–kol-bindningar måste bildas intill varandra i ett enda steg. Denna studie presenterar en ljust-driven metod som använder nickelkatalysatorer för att montera sådana tätt packade kolramverk med en noggrannhet som tidigare varit mycket svår att uppnå, vilket öppnar snabbare vägar till läkemedelsliknande molekyler.
Varför intilliggande kolbindningar är svåra
Många moderna läkemedel fungerar bäst när deras kolatomer är ordnade i ett specifikt tredimensionellt mönster, ungefär som en nyckel som är utskuren för att passa ett enda lås. Ett särskilt värdefullt mönster innebär två intilliggande kolatomer som vardera blir nya förgreningspunkter och förvandlar en plan kol–kol-dubbelbindning till en kompakt, mättad struktur. Interna alkener — dubbelbindningar inbäddade inne i en molekyl snarare än i slutet — är attraktiva utgångspunkter för detta, men de medför två problem. Deras kringliggande volym saktar ner den första radikaltillsatsen, och när det första nya fragmentet väl har bundit kan två liknande reaktiva kolfragment konkurrera på ett okontrollerat sätt, vilket gör det svårt att bestämma vilket fragment som hamnar var och på vilken sida av molekylen.
Använda ljus och nickel som ett samordnat team
Forskarna konstruerade en ”metallafotoredox”-plattform, där synligt ljus exciterar en fotokatalysator eller en speciell radikalkälla medan en nickelkomplex styr bindningsbildningen. Tillsammans genererar dessa katalysatorer kortlivade kolradikaler som adderar till interna alkener och sedan kanaliserar det resulterande intermediäret in i ett nickelbaserat kopplingssteg som fäster ett andra kolfragment. Genom att välja olika ligander — de små molekylerna bundna till nickel — kan de växla mellan två användbara lägen. Med en terpyridinligand och två alkylpartner utför systemet 1,2-dialkylation, vilket installerar två kolgrenar i en ”anti”-arrangemang. Med en kiral biimidazol-ligand och en alkyl plus ett arylfragment utförs 1,2-alkylarylering, återigen anti, men nu också selekterande för en enda spegelbildsform med hög precision.
Vad de nya reaktionerna kan bygga
Under mild blåljusbestrålning omvandlar dialkylationsprotokollet en rad cykliska och acykliska elektronfattiga interna alkener till produkter som bär två nya intilliggande C(sp³)–C-bindningar, ofta i hög avkastning och med utmärkt kontroll över vilken sida av dubbelbindningen varje fragment upptar. Metoden tolererar många funktionella grupper och fungerar till och med på komplexa naturliga produkter och fluorescerande prob- molekyler, vilket tillåter kemister att lägga till sp³-rika ”handtag” till molekyler sent i en syntes. Alkylaryleringsvarianten, som använder en kiral nickel–biimidazol-katalysator och en separat fotokatalysator, uppnår både hög diastereoselektivitet och enantioselektivitet. Den omvandlar kumariner, kinolinoner och besläktade ringar till β-aryl-α-alkyl-laktoner och liknande skelett som innehåller två intilliggande stereocenter — strukturer högt värderade inom medicinsk kemi.
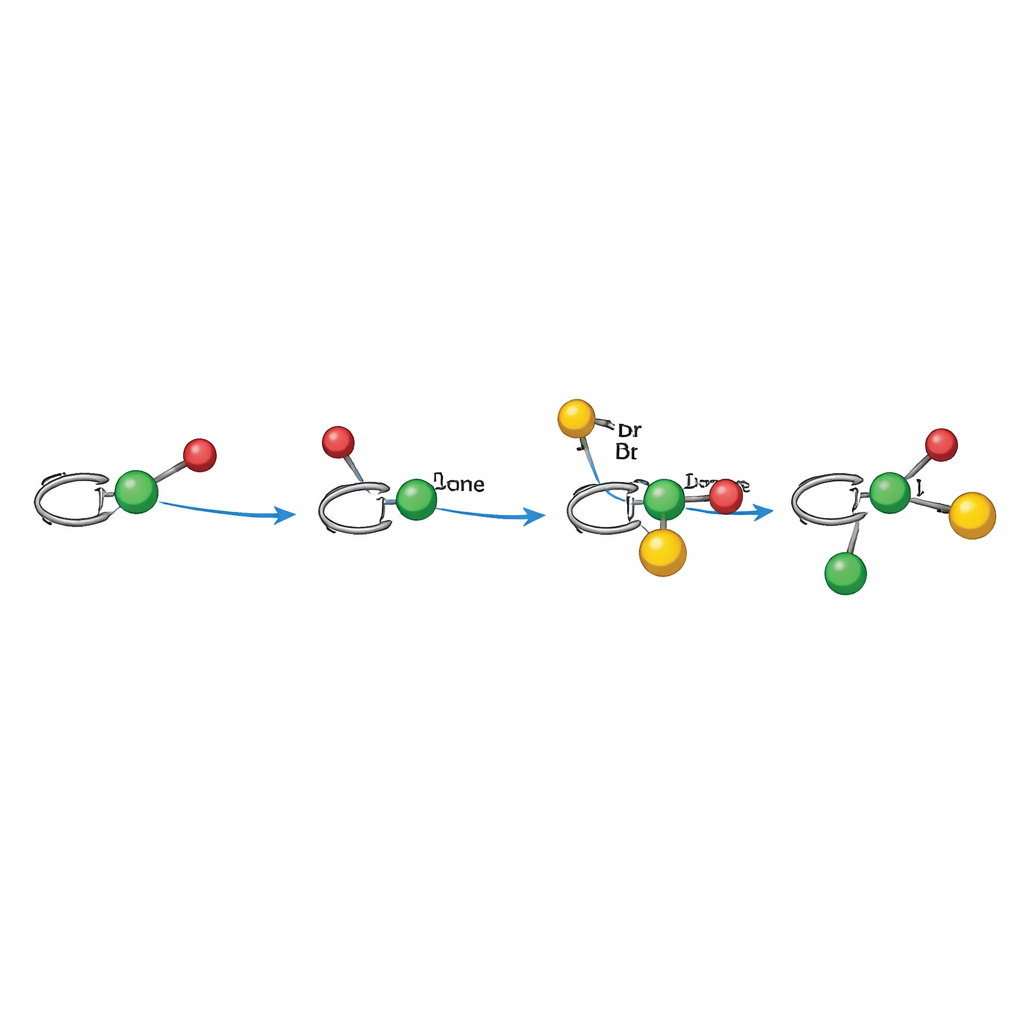
Hur den ljustriggade mekanismen fungerar
Mekanistiska experiment stöder en radikal väg som noggrant koreograferas av nickel. I alkylaryleringsläget aktiverar ljuset först fotokatalysatorn, som genererar en alkylradikal från en redox-aktiv ester. Den radikalen adderar till alkenen och bildar en benzylic radikal som är bunden — direkt eller indirekt — till ett kiralt nickelcentrum som redan engagerat arylbromiden. Nickelkomplexet fångar sedan den benzylica radikalen och formar den nya kol–kol-bindningen, frigör produkten och regenererar katalysatorn genom elektronöverföring. I dialkylationsläget tjänar en Hantzsch-ester dubbelt som radikalkälla och intern fotosensitiserare, reducerar direkt nickel och producerar en alkylradikal under ljus. I båda fallen styr ligandens form runt nickel vilken sida av alkenen som attackeras och hur de slutliga bindningarna sluts, vilket förklarar den starka kontrollen över tredimensionell ordning.
Vad detta betyder för framtida molekyler
Genom att förvandla en seg intern dubbelbindning till en port för två precist placerade kolgrenar ger detta arbete kemister en kraftfull genväg till komplexa, tredimensionella ramverk. De dubbla protokollen — en för rena alkylfragment och en för alkyl plus aryl — opererar under milda, reducerande förhållanden och tolererar många känsliga grupper, vilket gör dem särskilt lämpade för sent-stadie-modifiering av läkemedelskandidater. För en icke-specialist är huvudbudskapet att användning av ljus och nickel tillsammans skapar en finjusterad monteringslinje för att bygga trångbodda kolgrannskap som tidigare var svåra att nå, vilket potentiellt snabbar upp upptäckt och optimering av nya läkemedel.
Citering: Zhang, Y., Long, T., Sun, Y. et al. Stereoselective vicinal C(sp³)–C bond formation via metallaphotoredox 1,2-difunctionalization of internal alkenes. Nat Commun 17, 3066 (2026). https://doi.org/10.1038/s41467-026-69838-7
Nyckelord: radikal dicarbofunktionalisering, metallafotoredox-katalys, nickelkatalys, interna alkener, stereoselektiv C–C-bindningsbildning