Clear Sky Science · pl
Stereoselektywne tworzenie sąsiednich wiązań C(sp³)–C za pomocą metallafotoredoksowej 1,2-difunkcjonalizacji alkenów wewnętrznych
Budowanie złożonych cząsteczek za pomocą światła
Chemicy często muszą łączyć małe fragmenty węgla, aby uzyskać złożone trójwymiarowe struktury stosowane w lekach i materiałach. Zrobienie tego szybko, czysto i w kontrolowany sposób bywa zaskakująco trudne, szczególnie gdy w jednym kroku trzeba utworzyć dwa nowe wiązania węglowo‑węglowe obok siebie. W tym badaniu przedstawiono metodę napędzaną światłem, wykorzystującą katalizę niklową do składania gęsto upakowanych szkieletów węglowych z precyzją, która do tej pory była bardzo trudna do osiągnięcia, otwierając szybsze drogi do cząsteczek o charakterze lekowym.
Dlaczego sąsiednie wiązania węglowe są trudne do zbudowania
Wiele nowoczesnych leków działa najlepiej, gdy atomy węgla są ułożone w określonym trójwymiarowym porządku, podobnie jak klucz wycięty tak, żeby pasował do konkretnej zamki. Szczególnie cennym układem są dwa sąsiednie atomy węgla, które stają się punktami rozgałęzienia, przekształcając płaskie wiązanie podwójne w zwartą, nasyconą strukturę. Alkeny wewnętrzne — wiązania podwójne umieszczone wewnątrz cząsteczki, a nie na jej końcu — są atrakcyjnymi punktami wyjściowymi do tego typu transformacji, ale stawiają dwa problemy. Ich steryczne otoczenie spowalnia pierwsze przyłączenie rodnikowe, a po dodaniu pierwszego fragmentu dwa podobnie reaktywne fragmenty węglowe mogą konkurować w sposób niekontrolowany, co utrudnia decydowanie, który fragment przyłączy się gdzie i z której strony cząsteczki.
Wykorzystanie światła i niklu jako skoordynowanego zespołu
Naukowcy zaprojektowali platformę „metallafotoredoksową”, w której światło widzialne wzbudza fotokatalizator lub specjalne źródło rodnikowe, podczas gdy kompleks niklu kieruje tworzeniem wiązań. Razem te katalizatory generują krótkotrwałe rodniki węglowe, które addycjonują do alkenów wewnętrznych, a następnie kierują powstały intermediat do stadium sprzęgania kontrolowanego przez nikiel, które przyłącza drugi fragment węglowy. Dobierając różne ligandy — małe molekuły związane z niklem — można przełączać się między dwoma użytecznymi trybami. Z ligandem terpyrydynowym i dwoma partnerami alkilowymi system wykonuje 1,2‑dialkylację, instalując dwa rozgałęzienia w układzie „anti”. Z chiralnym ligandem biimidazolowym oraz jednym fragmentem alkilowym i jednym arylowym odbywa się 1,2‑alkilaryzacja, także w układzie anti, ale dodatkowo wybierana jest pojedyncza forma lustrzana z wysoką precyzją.
Co nowe reakcje potrafią zbudować
W łagodnych warunkach z użyciem niebieskiego światła protokół dialkylacji przekształca różne cykliczne i acykliczne, elektrono‑ubogie alkeny wewnętrzne w produkty zawierające dwa nowe sąsiednie wiązania C(sp³)–C, często w wysokich wydajnościach i z doskonałą kontrolą nad tym, która strona wiązania podwójnego otrzymuje dany fragment. Metoda toleruje wiele grup funkcyjnych i działa nawet na złożonych produktach naturalnych oraz sondach fluorescencyjnych, pozwalając chemikom dodać bogate w sp³ „uchwyty” do cząsteczek na późnym etapie syntezy. Wariant alkilaryzacji, stosujący chiralny kompleks nikiel–biimidazol oraz oddzielny fotokatalizator, osiąga zarówno wysoką diastereoselektywność, jak i enantioselektwyność. Przekształca kumaryny, chinolinony i pokrewne pierścienie w β‑arylowe‑α‑alkilowe laktony i podobne szkielety zawierające dwa sąsiednie centra stereogeniczne — struktury cenione w chemii medycznej.
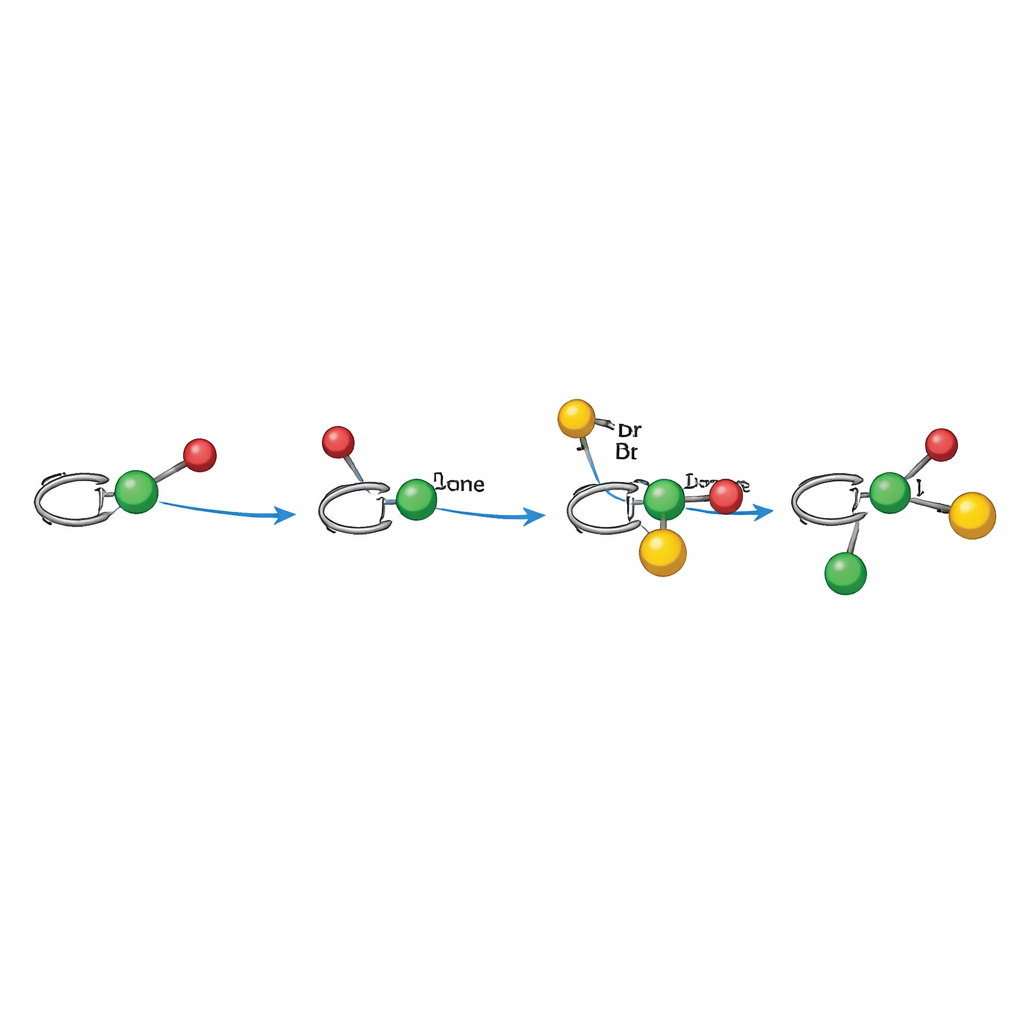
Jak działa mechanizm napędzany światłem
Eksperymenty mechanistyczne wspierają ścieżkę rodnikową skrupulatnie ukierunkowaną przez nikiel. W trybie alkilaryzacji światło najpierw aktywuje fotokatalizator, który generuje rodnik alkilowy z estru aktywnego redoksowo. Ten rodnik przyłącza się do alkenu, tworząc rodnik benzyliczny, który jest związany — bezpośrednio lub pośrednio — z chiralnym centrum niklu, które już weszło w reakcję z bromkiem arylowym. Kompleks niklu następnie wychwytuje rodnik benzyliczny i tworzy nowe wiązanie węglowo‑węglowe, uwalniając produkt i regenerując katalizator przez transfer elektronowy. W trybie dialkylacji ester Hantzscha pełni podwójną rolę jako źródło rodnika i wewnętrzny fotosensybilizator, bezpośrednio redukując nikiel i produkując rodnik alkilowy pod wpływem światła. W obu przypadkach kształt ligandu wokół niklu steruje, która strona alkenu jest atakowana i jak zamykają się ostateczne wiązania, co wyjaśnia silną kontrolę nad ułożeniem trójwymiarowym.
Co to oznacza dla przyszłych cząsteczek
Przekształcając oporne wiązanie podwójne wewnętrzne w bramę do wprowadzenia dwóch precyzyjnie umieszczonych rozgałęzień węglowych, ta praca oferuje chemikom potężny skrót do złożonych, trójwymiarowych szkieletów. Oba protokoły — jeden dla fragmentów czysto alkilowych, drugi dla alkilów i arylów — działają w łagodnych, redukcyjnych warunkach i tolerują wiele wrażliwych grup, dzięki czemu są szczególnie przydatne do modyfikacji kandydatów na leki na późnym etapie. Dla laika kluczowa informacja jest taka: użycie światła i niklu razem tworzy precyzyjną linię montażową do budowy zatłoczonych środowisk węglowych, które wcześniej były trudne do osiągnięcia, co może przyspieszyć odkrywanie i optymalizację nowych leków.
Cytowanie: Zhang, Y., Long, T., Sun, Y. et al. Stereoselective vicinal C(sp³)–C bond formation via metallaphotoredox 1,2-difunctionalization of internal alkenes. Nat Commun 17, 3066 (2026). https://doi.org/10.1038/s41467-026-69838-7
Słowa kluczowe: rodnikowa dikarbofunkcjonalizacja, kataliza metallafotoredoksowa, kataliza niklowa, alkeny wewnętrzne, stereoselektywne tworzenie wiązań C–C