Clear Sky Science · en
Stereoselective vicinal C(sp³)–C bond formation via metallaphotoredox 1,2-difunctionalization of internal alkenes
Building Complex Molecules with Light
Chemists often need to stitch together small carbon fragments into intricate three-dimensional shapes for medicines and materials. Doing this quickly, cleanly, and in a controlled way is surprisingly hard, especially when two new carbon–carbon links must be formed next to each other in a single step. This study presents a light-driven method that uses nickel catalysts to assemble such densely packed carbon frameworks with a level of precision that has been very difficult to achieve, opening faster routes to drug-like molecules.
Why Neighboring Carbon Links Are Tough
Many modern drugs work best when their carbon atoms are arranged in a specific three-dimensional pattern, much like a key cut to fit one lock. A particularly valuable pattern involves two neighboring carbon atoms that each become new branching points, turning a flat carbon–carbon double bond into a compact, saturated structure. Internal alkenes—double bonds tucked inside a molecule rather than at the end—are attractive starting points for this, but they pose two problems. Their bulk slows the first radical addition, and once the first new piece has attached, two similarly reactive carbon fragments can compete in an uncontrolled way, making it hard to decide which piece goes where and on which face of the molecule.
Using Light and Nickel as a Coordinated Team
The researchers designed a “metallaphotoredox” platform, in which visible light excites a photocatalyst or a special radical source while a nickel complex guides bond formation. Together, these catalysts generate short-lived carbon radicals that add to internal alkenes, then channel the resulting intermediate into a nickel-based coupling step that fixes a second carbon fragment. By choosing different ligands—the small molecules bound to nickel—they can switch between two useful modes. With a terpyridine ligand and two alkyl partners, the system performs 1,2-dialkylation, installing two carbon branches in an “anti” arrangement. With a chiral biimidazole ligand and one alkyl plus one aryl fragment, it performs 1,2-alkylarylation, again anti, but now also selecting a single mirror-image form with high precision.
What the New Reactions Can Build
Under mild blue-light irradiation, the dialkylation protocol converts a variety of cyclic and acyclic electron-poor internal alkenes into products bearing two new neighboring C(sp³)–C bonds, often in high yield and with excellent control over which side of the double bond each fragment occupies. The method tolerates many functional groups and even works on complex natural products and fluorescent probes, allowing chemists to add sp³-rich “handles” to molecules late in a synthesis. The alkylarylation variant, using a chiral nickel–biimidazole catalyst and a separate photocatalyst, achieves both high diastereoselectivity and enantioselectivity. It transforms coumarins, quinolinones, and related rings into β-aryl-α-alkyl lactones and similar scaffolds that contain two adjacent stereocenters, structures prized in medicinal chemistry.
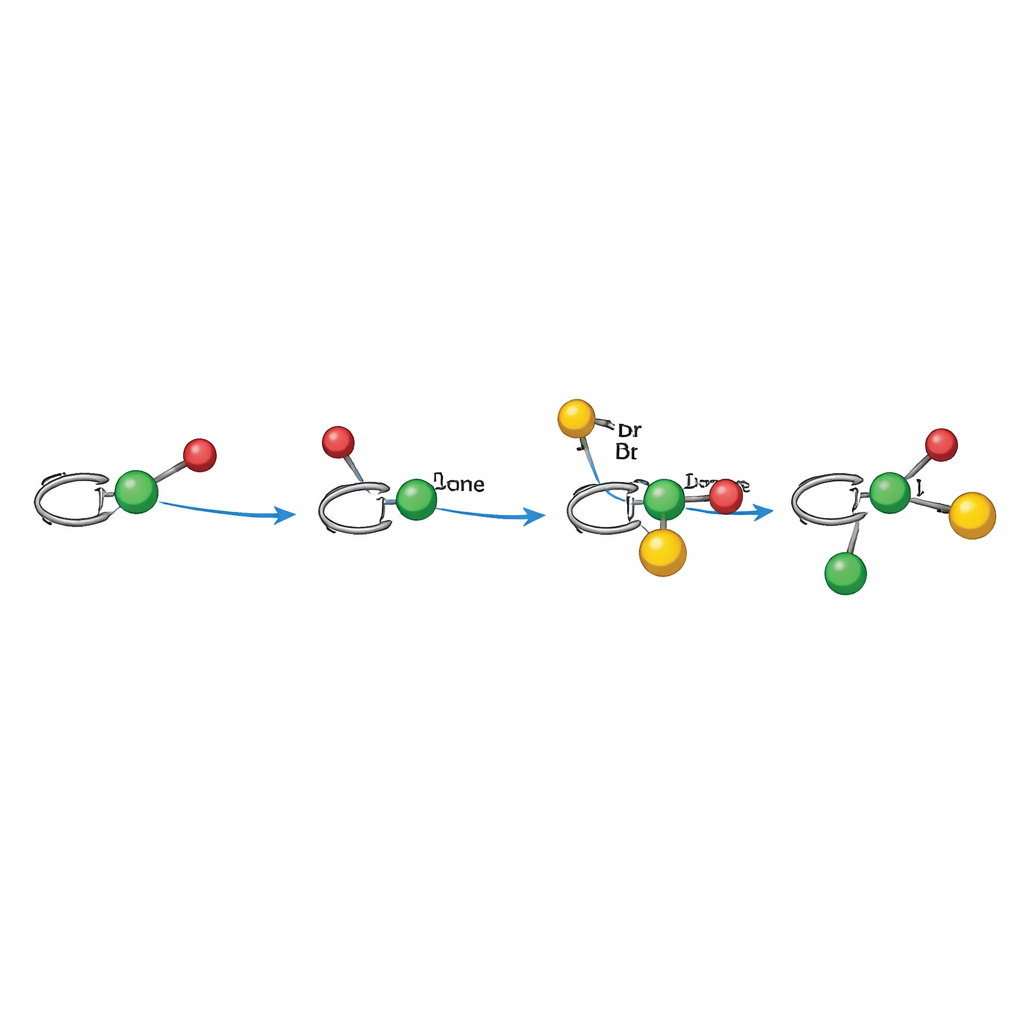
How the Light-Driven Mechanism Works
Mechanistic experiments support a radical pathway carefully choreographed by nickel. In the alkylarylation mode, light first activates the photocatalyst, which generates an alkyl radical from a redox-active ester. That radical adds to the alkene, forming a benzylic radical that is bound—directly or indirectly—to a chiral nickel center that has already engaged the aryl bromide. The nickel complex then captures the benzylic radical and forges the new carbon–carbon bond, releasing the product and regenerating the catalyst through electron transfer. In the dialkylation mode, a Hantzsch ester double-serves as radical source and internal photosensitizer, directly reducing nickel and producing an alkyl radical under light. In both cases, the ligand shape around nickel steers which face of the alkene is attacked and how the final bonds close, explaining the strong control over three-dimensional arrangement.
What This Means for Future Molecules
By turning a stubborn internal double bond into a gateway for two precisely placed carbon branches, this work offers chemists a powerful shortcut to complex, three-dimensional frameworks. The dual protocols—one for purely alkyl fragments and one for alkyl plus aryl—operate under gentle, reducing conditions and tolerate many sensitive groups, making them especially suited for late-stage tweaking of drug candidates. For a non-specialist, the take-home message is that using light and nickel together provides a finely tuned assembly line for building crowded carbon neighborhoods that were previously difficult to access, potentially speeding the discovery and optimization of new medicines.
Citation: Zhang, Y., Long, T., Sun, Y. et al. Stereoselective vicinal C(sp³)–C bond formation via metallaphotoredox 1,2-difunctionalization of internal alkenes. Nat Commun 17, 3066 (2026). https://doi.org/10.1038/s41467-026-69838-7
Keywords: radical dicarbofunctionalization, metallaphotoredox catalysis, nickel catalysis, internal alkenes, stereoselective C–C bond formation