Clear Sky Science · nl
Stereoselectieve vicinale C(sp³)–C-bindingvorming via metallaphotoredox 1,2-difunctionalisatie van interne alkenen
Complexe moleculen bouwen met licht
Chemici moeten vaak kleine koolstoffragmenten aan elkaar rijgen tot ingewikkelde driedimensionale vormen voor geneesmiddelen en materialen. Dat snel, schoon en gecontroleerd doen is verrassend moeilijk, vooral wanneer in één stap twee nieuwe koolstof–koolstofverbindingen naast elkaar moeten worden gevormd. Deze studie presenteert een lichtgestuurde methode die nikkelkatalysatoren gebruikt om zulke dicht bezette koolstofkaders samen te stellen met een precisie die tot nu toe zeer moeilijk te bereiken was, en opent daarmee snellere routes naar geneesmiddelachtige moleculen.
Waarom aangrenzende koolstofverbindingen lastig zijn
Veel moderne geneesmiddelen werken het beste wanneer hun koolstofatomen in een specifiek driedimensionaal patroon zijn gerangschikt, vergelijkbaar met een sleutel die op maat is geslepen voor één slot. Een bijzonder waardevol patroon omvat twee aangrenzende koolstofatomen die elk nieuwe vertakkingspunten worden, waarbij een vlakke koolstof–koolstof-dubbele binding verandert in een compacte, verzadigde structuur. Interne alkenen—dubbele bindingen die in het molecuul zijn ingebed in plaats van aan het uiteinde—zijn aantrekkelijke uitgangspunten hiervoor, maar ze vormen twee problemen. Hun volumineuze omgeving vertraagt de eerste radicalaire additie, en zodra het eerste nieuwe fragment is aangehecht kunnen twee vergelijkbaar reactieve koolstoffragmenten ongecontroleerd met elkaar concurreren, waardoor het moeilijk is te bepalen welk fragment waar en aan welke zijde van het molecuul wordt geplaatst.
Licht en nikkel als een gecoördineerd team
De onderzoekers ontwierpen een "metallaphotoredox" platform, waarbij zichtbaar licht een fotokatalysator of een speciale radicale bron exciteert terwijl een nikkelcomplex de vorming van bindingen begeleidt. Gezamenlijk genereren deze katalysatoren kortlevende koolstofradicalen die additie aan interne alkenen ondergaan en het resulterende intermediair vervolgens in een nikkel-gedragen koppeling sturen die een tweede koolstoffragment bevestigt. Door verschillende liganden te kiezen—de kleine moleculen gebonden aan nikkel—kunnen ze schakelen tussen twee nuttige modi. Met een terpyridineligand en twee alkylpartners voert het systeem 1,2-dialkylatie uit en installeert twee koolstofvertakkingen in een "anti"-arrangement. Met een chiraal biimidazool-ligand en één alkyl plus één arylfragment voert het 1,2-alkylarylering uit, eveneens anti, maar nu ook met hoge selectie van één spiegelbeeldvorm.
Wat de nieuwe reacties kunnen bouwen
Onder milde blauwlichtbestraling zet het dialkylatieprotocol een verscheidenheid aan cyclische en acyclische elektronarme interne alkenen om in producten met twee nieuwe aangrenzende C(sp³)–C-bindingen, vaak met hoge opbrengst en uitstekende controle over aan welke zijde van de dubbele binding elk fragment zit. De methode verdraagt veel functionele groepen en werkt zelfs op complexe natuurlijke producten en fluorescerende probes, waardoor chemici sp³-rijke "handvatten" laat in een synthese aan moleculen kunnen toevoegen. De alkylaryleringsvariant, met een chiraal nikkel–biimidazool-katalysator en een aparte fotokatalysator, bereikt zowel hoge diastereoselectiviteit als enantioselectiviteit. Ze transformeert coumarines, quinolinonen en verwante ringen in β-aryl-α-alkyl-lactonen en soortgelijke skeletstructuren die twee aangrenzende stereocentra bevatten, structuren die zeer gewild zijn in de medische chemie.
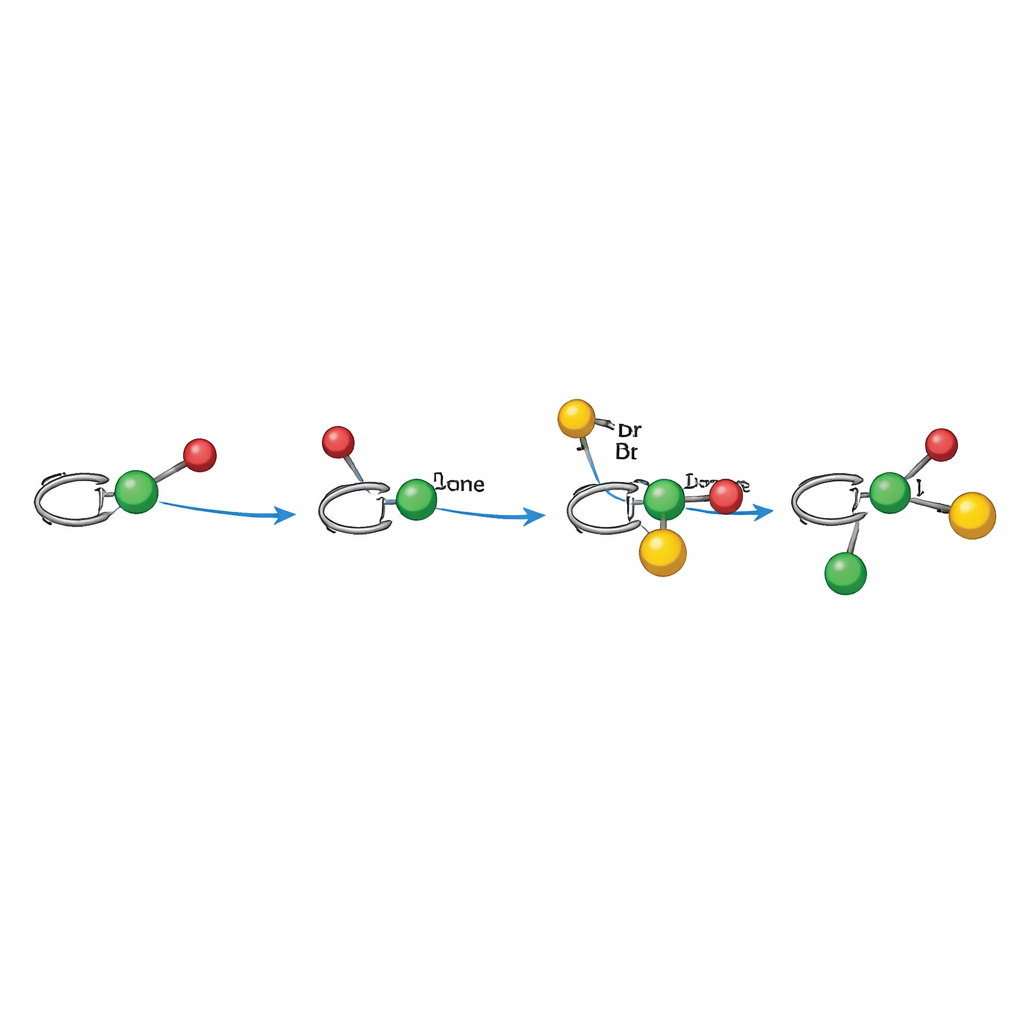
Hoe het lichtgestuurde mechanisme werkt
Mechanistische experimenten ondersteunen een radicaal pad dat zorgvuldig door nikkel wordt gecorrigeerd. In de alkylaryleringsmodus activeert licht eerst de fotokatalysator, die een alkylradicaal genereert uit een redox-actief ester. Dat radicaal voegt zich bij de alkeen en vormt een benzylic-radicaal dat rechtstreeks of indirect wordt gebonden aan een chiraal nikkelcentrum dat het arylbromide al heeft aangegaan. Het nikkelcomplex vangt vervolgens het benzylic-radicaal op en vormt de nieuwe koolstof–koolstofbinding, waarbij het product vrijkomt en de katalysator door elektronenoverdracht wordt geregenereerd. In de dialkylatiemodus dient een Hantzsch-ester dubbel als radicale bron en interne fotosensitizer, reduceert rechtstreeks nikkel en produceert onder licht een alkylradicaal. In beide gevallen stuurt de ligandvorm rondom nikkel welke zijde van de alkeen wordt aangevallen en hoe de uiteindelijke bindingen sluiten, wat de sterke controle over de driedimensionale ordening verklaart.
Wat dit betekent voor toekomstige moleculen
Door een hardnekkige interne dubbele binding te veranderen in een toegangspoort voor twee precies geplaatste koolstofvertakkingen, biedt dit werk chemici een krachtig kortere route naar complexe, driedimensionale kaders. De twee protocollen—één voor puur alkylfragmenten en één voor alkyl plus aryl—werken onder milde, reducerende omstandigheden en verdragen vele gevoelige groepen, waardoor ze bijzonder geschikt zijn voor laat-stadium aanpassingen van kandidaat-geneesmiddelen. Voor niet-specialisten is de kernboodschap dat het combineren van licht en nikkel een fijn afgestelde assemblagelijn biedt om drukbevolkte koolstofbuurten te bouwen die voorheen moeilijk toegankelijk waren, wat potentieel de ontdekking en optimalisatie van nieuwe geneesmiddelen kan versnellen.
Bronvermelding: Zhang, Y., Long, T., Sun, Y. et al. Stereoselective vicinal C(sp³)–C bond formation via metallaphotoredox 1,2-difunctionalization of internal alkenes. Nat Commun 17, 3066 (2026). https://doi.org/10.1038/s41467-026-69838-7
Trefwoorden: radicale dicarbofunctionalisatie, metallaphotoredox katalyse, nikkel katalyse, interne alkenen, stereoselectieve C–C-bindingvorming